{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
This website contains information regarding the paper Modular Flows: Differential Molecular Generation.
TL;DR: We propose generative graph normalizing flow models, based on a system of coupled node ODEs, that repeatedly reconcile locally toward globally aligned densities for high quality molecular generation
Please cite our work if you find it useful:
@misc{https://doi.org/10.48550/arxiv.2210.06032,
doi = {10.48550/ARXIV.2210.06032},
url = {https://arxiv.org/abs/2210.06032},
author = {Verma, Yogesh and Kaski, Samuel and Heinonen, Markus and Garg, Vikas},
keywords = {Machine Learning (cs.LG), Emerging Technologies (cs.ET), Biomolecules (q-bio.BM), Machine Learning (stat.ML), FOS: Computer and information sciences, FOS: Computer and information sciences, FOS: Biological sciences, FOS: Biological sciences},
title = {Modular Flows: Differential Molecular Generation},
publisher = {arXiv},
year = {2022},
copyright = {Creative Commons Attribution 4.0 International}
}
Generating new molecules is fundamental to advancing critical applications such as drug discovery and material synthesis. A key challenge of molecular generative models is to be able to generate valid molecules, according to various criteria for molecular validity or feasibility. It is a common practice to use external chemical software as rejection oracles to reduce or exclude invalid molecules, or do validity checks as part of autoregressive generation [1,2,3] . An important open question has been whether generative models can learn to achieve high generative validity intrinsically, i.e., without being aided by oracles or performing additional checks. We circumvent the issues with novel physics-inspired co-evolving continuous-time flows that induces useful inductive biases for a highly complex combinatorial setting. Our method is inspired by graph PDEs, that repeatedly reconcile locally toward globally aligned densities.
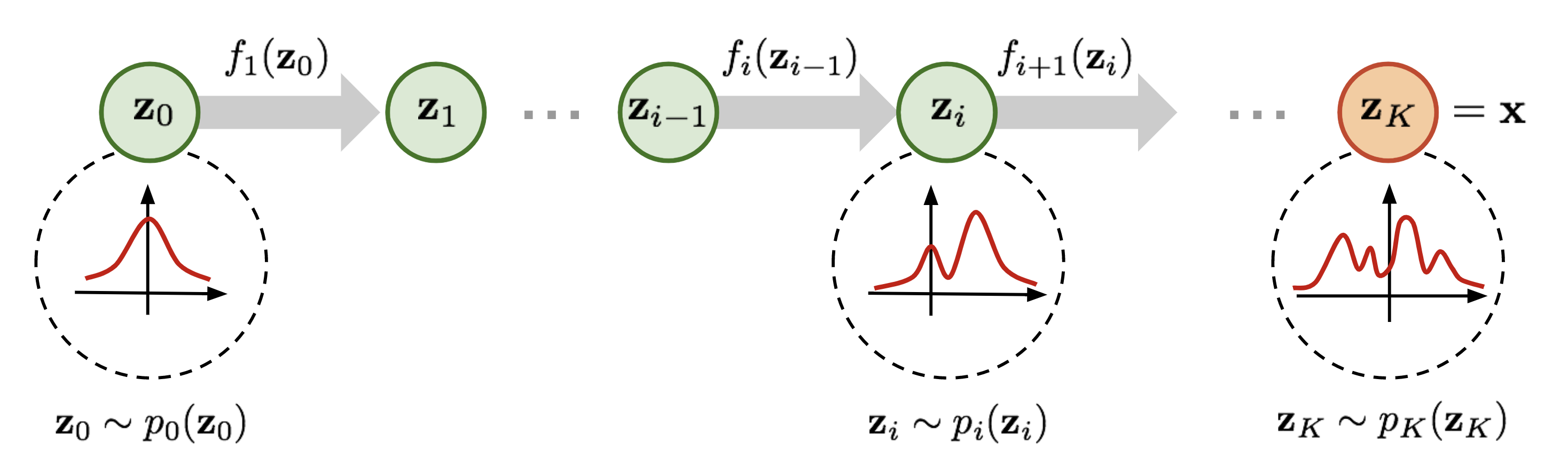
Normalizing flow have seen widespread use for density modeling, generative modeling, etc which provides a general way of constructing flexible probability distributions. It is defined by a parameterized invertible deterministic transformation from a base distribution
Given a datapoint
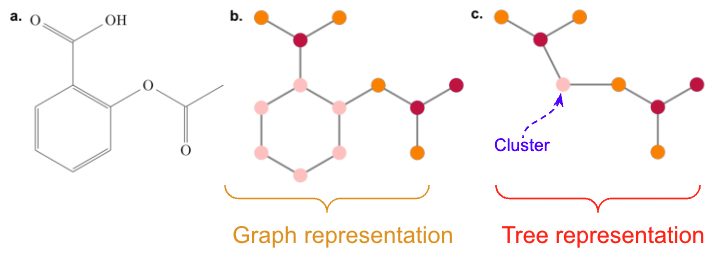
We represent molecule as a graph
We can obtain an alternative representation by decomposing a moleculer graph into a tree, by contracting certain vertices into a single node such that the molecular graph
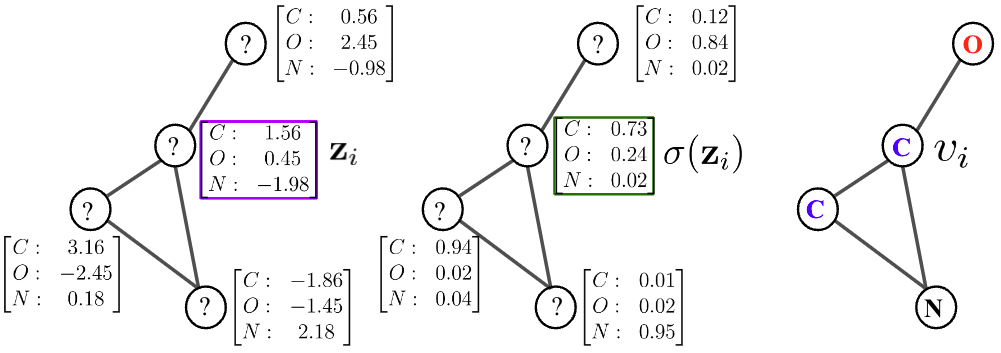
Based on the general recipie of normalizing flows, we propose to model the node scores $$\mathbf{z}{i}$$ as a Continuous-time Normalizing Flow (CNF)[7] over time $$t \in \mathrm{R}+$$. We assume the initial scores at time
where $$\mathcal{N}{i} = { \mathbf{z}{j} : (i,j) \in E }$$ is the set of neighbor scores at time
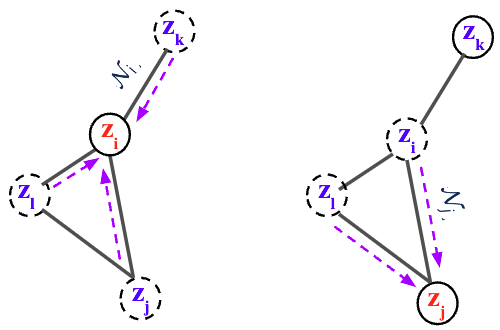
By collecting all node differentials we obtain a modular joint, coupled ODE, which is equivalent to a graph PDE [9,10], where the evolution of each node only depends on its immediate neighbors.
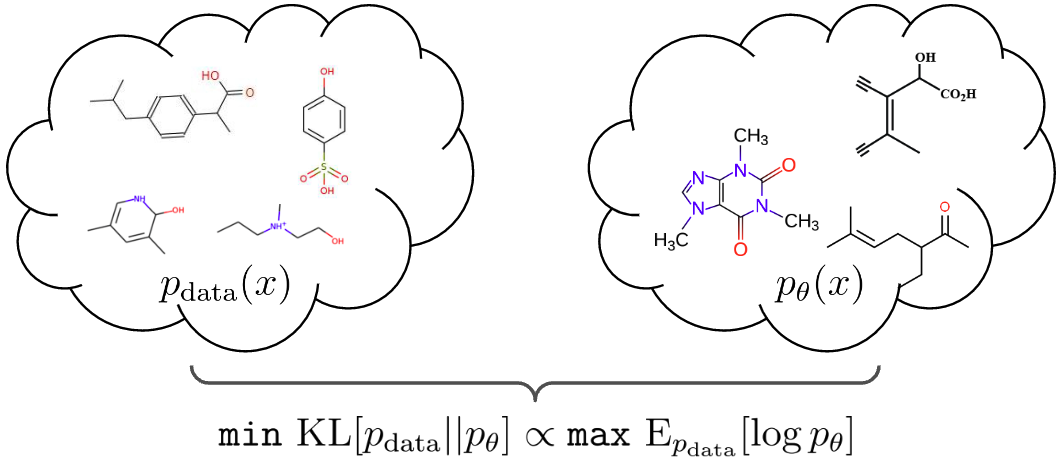
We reduce the learning problem to maximizing the score cross-entropy $$\mathrm{E}{\hat{p}{\mathrm{data}}(\mathbf{z}(T))}[\log p_\theta(\mathbf{z}(T))]$$, where we turn the observed set of graphs
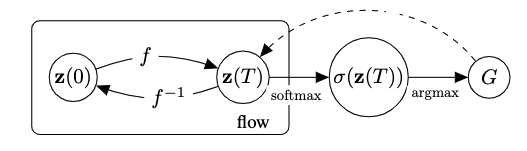
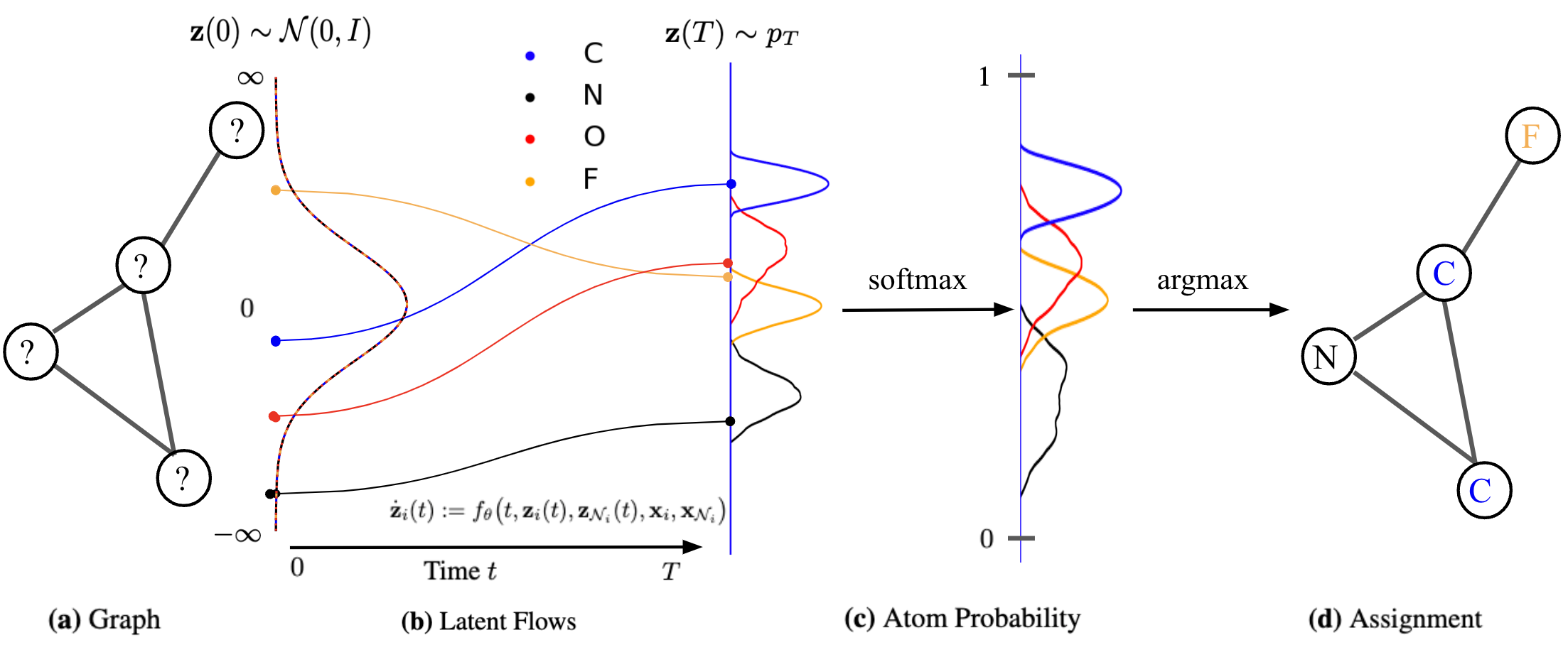
We exploit the non-reversible composition of the argmax and softmax to transition from continous space to discrete graph space, but short-circuit in reverse direction as shown in the figure below. This indeed allows to keep the forward and backward flows aligned. We thus maximize an objective over
We generate novel molecules by sampling an initial state
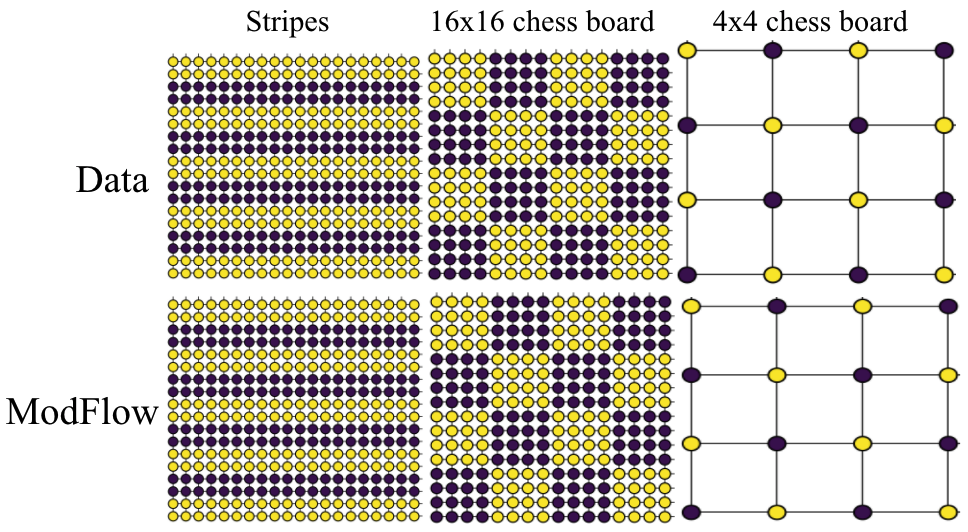
We demonstrated the power of our method on learning highly discontinous patterns on 2D grid graphs. We considered patterns corresponding to two-variants of chess-board pattern as
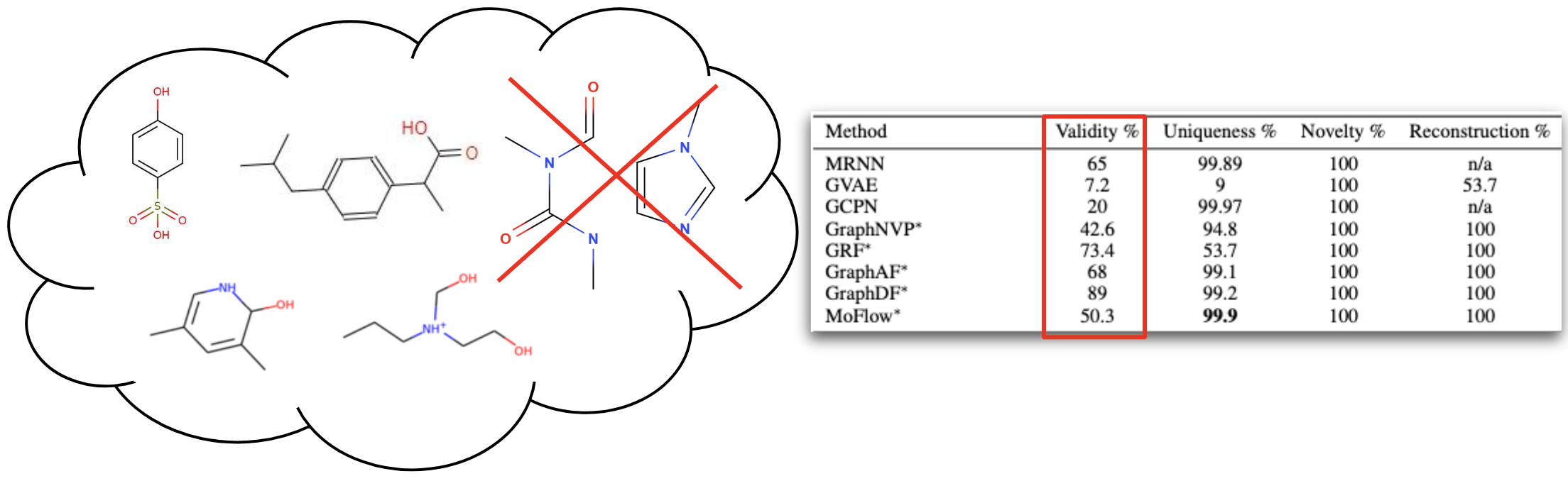
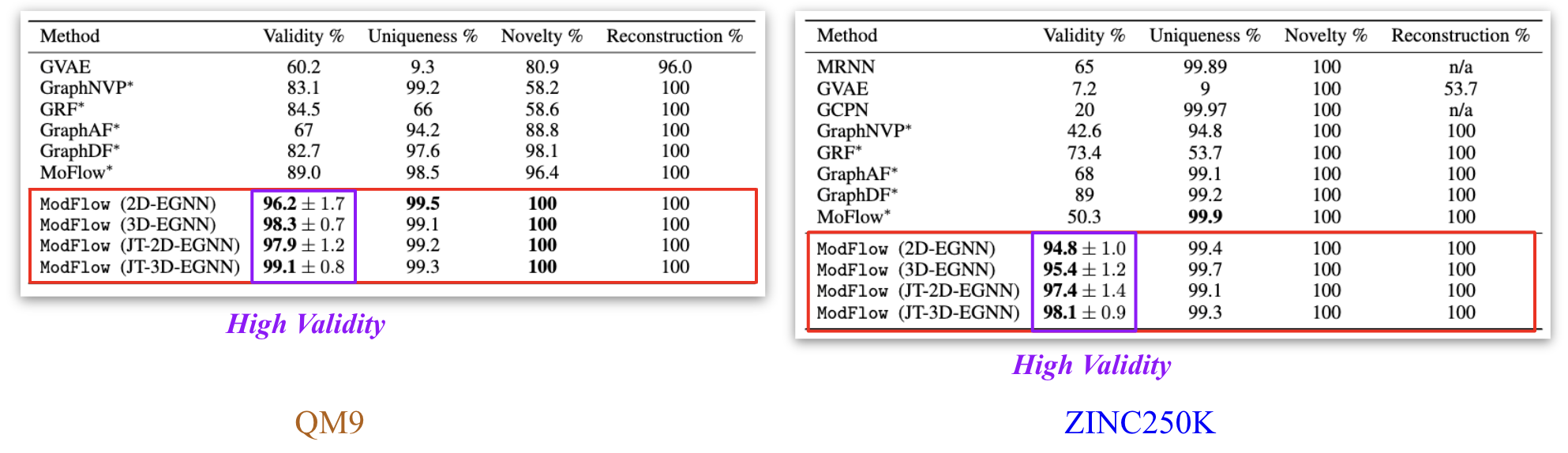
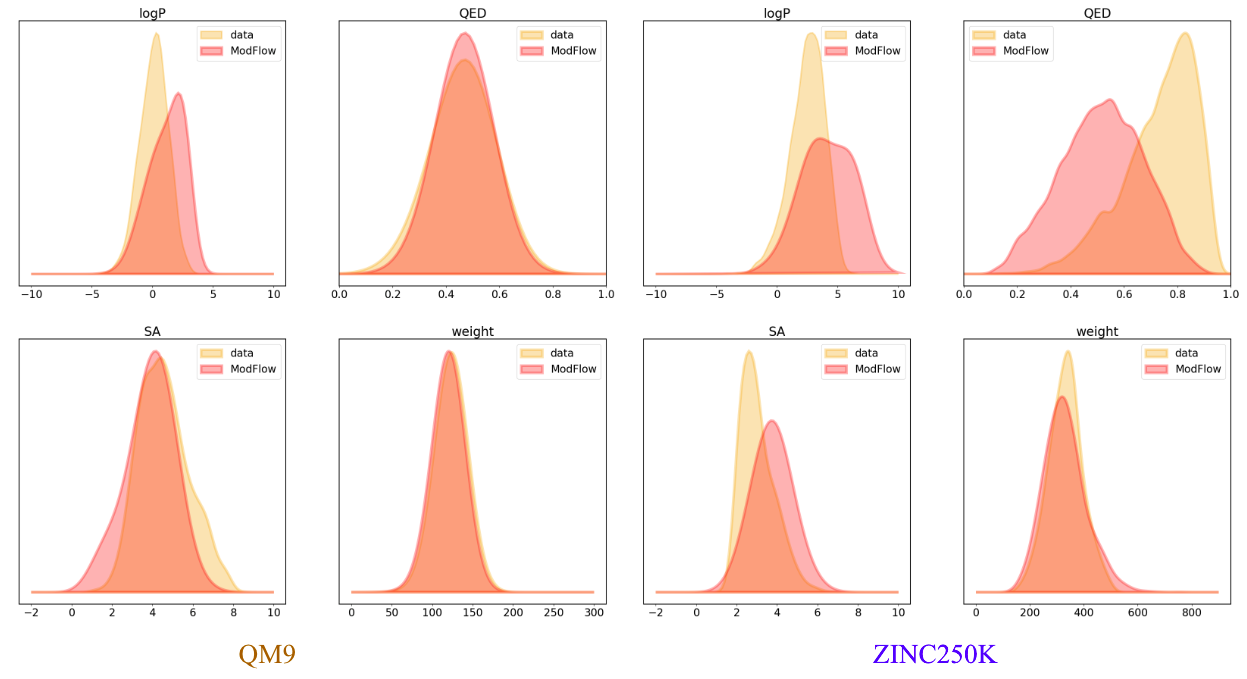
We trained the model on QM9[6] and ZINC250K[5] dataset, where molecules are in kekulized form with hydrogens removed by the RDkit[8] software. We adopt common quality metrics to evaluate molecular generation as,
- Validity: Fraction of molecules that satisfy chemical valency rule
- Uniqueness: Fraction of non-duplicate generations
- Novelty: Fraction of molecules not present in training data
- Reconstruction: Fraction of molecules that can be reconstructed from their encoding
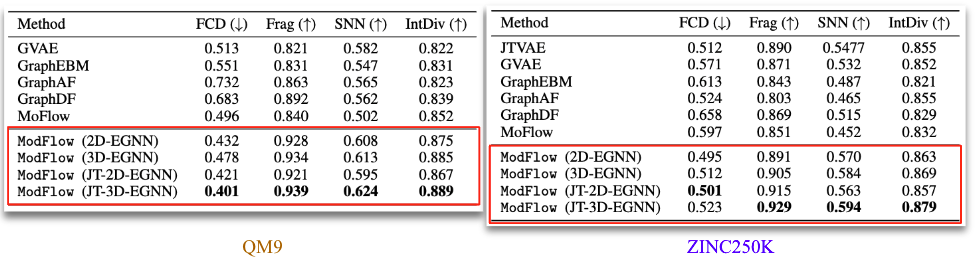
- FCD: measures diversity and chemical and biological property alignment
- SNN: quantifies closeness of generated molecules to true molecule manifold
- Frag: measures distance between the fragment frequencies generated and reference
- IntDiv: diversity by computing pairwise similarity of the generated molecules
Some of the generated molecules via
- Molecular Weight: Sum of the individual atomic weights of a molecule.
- LogP: Ratio of concentration in octanol-phase to aqueous phase, also known as the octanol-water partition coefficient.
- Synthetic Accessibility Score (SA): Estimate describing the synthesizability of a given molecule
- Quantitative Estimation of Drug-likeness (QED): Value describing likeliness of a molecule as a viable candidate for a drug
We performed Property-targeted Molecular Optimization, to search for molecules, having a better chemical properties. Specifically, we choose quantitative estimate of drug-likeness (QED) as our target chemical property, which measures the potential of a molecule to be characterized as a drug. We used a pre-trained ModFlow model
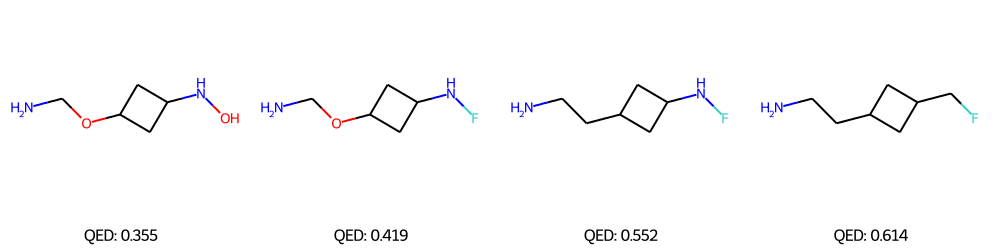
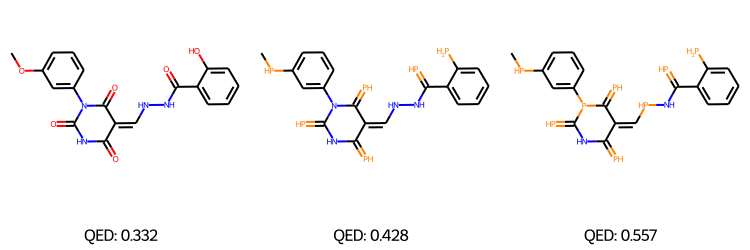
The above figures represent the molecules decoded from the learned latent space with linear regression for successful molecular optimization.
We performed ablation experiments to gain further insights about
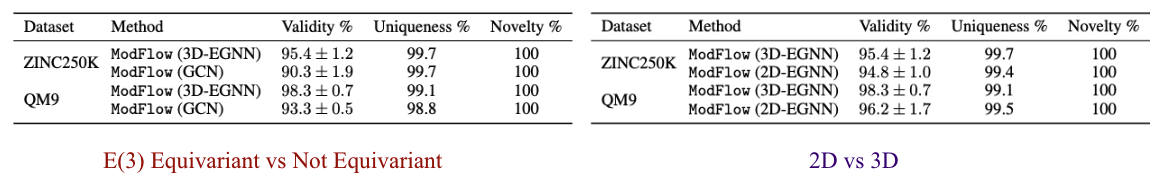
- We propose Physics-inspired co-evolving continuous-time flows, inspired by graph PDEs as
$$\texttt{ModFlow}$$ , where multiple flows interact locally according to a modular coupled ODE system.- The coupled dynamics results in accurate modeling of graph densities and high quality molecular generation without any validity checks or correction.
- Interesting avenues open up, including the design of (a) more nuanced mappings between discrete and continuous spaces, and (b) extensions of modular flows to (semi-)supervised settings.
- Youzhi Luo, Keqiang Yan, and Shuiwang Ji. Graphdf: A discrete flow model for molecular graph generation,2021
- Chence Shi, Minkai Xu, Zhaocheng Zhu, Weinan Zhang, Ming Zhang, and Jian Tang. Graphaf: a flow-based autoregressive model for molecular graph generation
- Mariya Popova, Mykhailo Shvets, Junier Oliva, and Olexandr Isayev. Molecularrnn: Generating realistic molecular graphs with optimized properties,2019
- Wengong Jin, Regina Barzilay, and Tommi Jaakkola. Junction tree variational autoencoder for molecular graph generation
- John J Irwin, Teague Sterling, Michael M Mysinger, Erin S Bolstad, and Ryan G Coleman. Zinc: a free tool to discover chemistry for biology. Journal of chemical information and modeling, 52(7):1757–1768, 2012
- Raghunathan Ramakrishnan, Pavlo O Dral, Matthias Rupp, and O Anatole von Lilienfeld. Quantum chemistry structures and properties of 134 kilo molecules. Scientific Data, 1, 2014
- Will Grathwohl, Ricky TQ Chen, Jesse Bettencourt, Ilya Sutskever, and David Duvenaud. Ffjord: Free-form continuous dynamics for scalable reversible generative models
- Greg Landrum et al. Rdkit: A software suite for cheminformatics, computational chemistry, and predictive modeling, 2013
- Valerii Iakovlev, Markus Heinonen, and Harri Lähdesmäki. Learning continuous-time pdes from sparse data with graph neural networks.
- Ben Chamberlain, James Rowbottom, Maria I Gorinova, Michael Bronstein, Stefan Webb, and Emanuele Rossi Grand: Graph neural diffusion. In International Conference on Machine Learning, pages 1407–1418. PMLR, 2021
- Victor Garcia Satorras, Emiel Hoogeboom, and Max Welling. E(n) equivariant graph neural networks, 2021