Extra what about that long read
We could note that some Nanopore reads for phage T7 were longer than the whole phage:
┌─────────────────┬───────┬──────────┬─────────┬───────┬───────┬───────┬───────────┬───────┬───────┐
│ File │ #Seq │ Total bp │ Avg │ N50 │ N75 │ N90 │ auN │ Min │ Max │
├─────────────────┼───────┼──────────┼─────────┼───────┼───────┼───────┼───────────┼───────┼───────┤
│ T7.fasta │ 1 │ 39778 │ 39778.0 │ 39778 │ 39778 │ 39778 │ 39778.000 │ 39778 │ 39778 │
│ T7-ONT.fastq.gz │ 51848 │ 77898476 │ 1502.4 │ 5768 │ 1717 │ 424 │ 7225.658 │ 112 │ 64864 │
└─────────────────┴───────┴──────────┴─────────┴───────┴───────┴───────┴───────────┴───────┴───────┘
There are several explanations: a couple of molecules from the host genome might have slipped through, for example. But we are curious and we want to dig further.
The whole genome is 40Kb, let us select the reads longer than 40Kb.
We can use SeqFu cat for this, and first we can check how many we have:
seqfu cat --min-len 40000 /data/reads/ont/T7-ONT.fastq.gz | seqfu countThe output is just 5, it's a reasonable number so we can check the exact length of each with asking SeqFu to add the length to the name, rename the reads to make the output easier to read and then "selecting" the headers with grep.
seqfu cat --min-len 40000 --prefix seq --add-len \
--fasta --strip-name --strip-comment /data/reads/ont/T7-ONT.fastq.gz | grep ">"Here we ask seqfu to:
- Select reads longer than 40Kb with
--min-len 40000 - Remove the original name and comments from the reads (they are very looong!) with
--strip-name --strip-comment - To rename the reads with a prefix with
--prefix seq - To print the output in FASTA (originally is FASTQ) with
--fasta - To append the read length to each read with
--add-len
The output then is filtered to only print the headers and not the sequences. The output is:
>seq_1 len=64864
>seq_2 len=40047
>seq_3 len=40003
>seq_4 len=43305
>seq_5 len=40232
There are 3 40kb sequences. They are likely the phage with some homopolymer errors. Let's extract the first sequence with a higher threshold and save it to a file.
We can then use "Blast" to compare the sequence with the rotated assembly we produced yesterday, using the "Blast two sequences" option: We can paste the two assemblies in Blast using the "Blast two sequences" options
The output also shows a "dot plot"
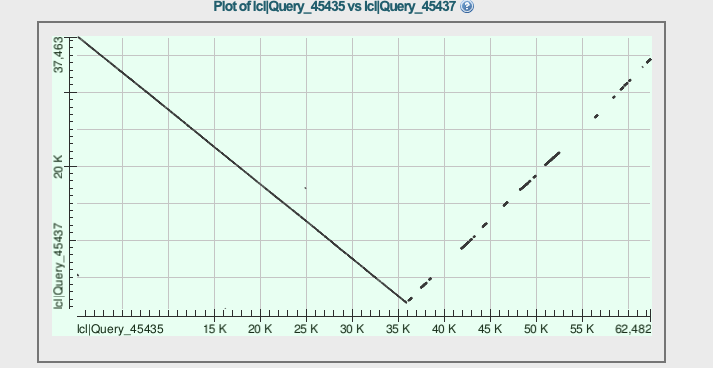
Apparently we have the whole phages from the beginning of the read, and then we have fragments of the phage in reverse direction. Something funny happened in that nanopore.
The Nanopore library was prepared with the "ligation" kit, and this phenomenon was already reported with LSK-109 ligation kit: