Extra alignment and coverage
We will use bwa to align short reads (while minimap2 is a good option for nanopore reads). Let's start creating an environment with sometools, and samtools ;)
# Create the environment called "align", this is just to be done once
mamba create -n align bwa minimap2 samtools bedtools bamtocov
# Activate the environment to use the tools
conda activate alignbwa is a fast and easy to use short reads mapper.
First you need to "index" the reference used to map the reads against. This step has to be done only the first time, then you can align multiple times.
# The syntax is "bwa index FASTA_REF"
bwa index ~/assembly/T4.fasta Then we can map the paired end reads. The output is in SAM format, a simple text format used to store the mapping positions and attributes. Short reads are usually a lot, so there is a compressed and optimized counterpart called "BAM" (Binary SAM), and a Swiss-army penknife to manipulate such files called samtools.
Here we use the power of the Unix pipes to stream the SAM file from BWA to samtools in order to convert it to a BAM, and to sort it by position. An "index file" can make the life of a lot of programs easier, so with samtools index we can easily create one for the freshly minted BAM file.
# Create a new directory
mkdir aln
# Perform the alignment and BAM sorting in a stream!
bwa mem ~/assembly/T4.fasta /data/reads/illumina/T4_R* | samtools view -bS | samtools sort -o aln/T4.bam
# index the BAM file
samtools index aln/T4.bam
# Check what you just created
ls -l aln/Mappings can be easily visualized having the reference sequence used to generate them, the BAM file (.bam) and it's associated index (.bam.bai).
You can use UGENE or the lighter IGV viewer.
In IGV from "Genome" select "Load genome..." and select the FASTA file, and then from "File" select "Load from file..." and select the BAM file:
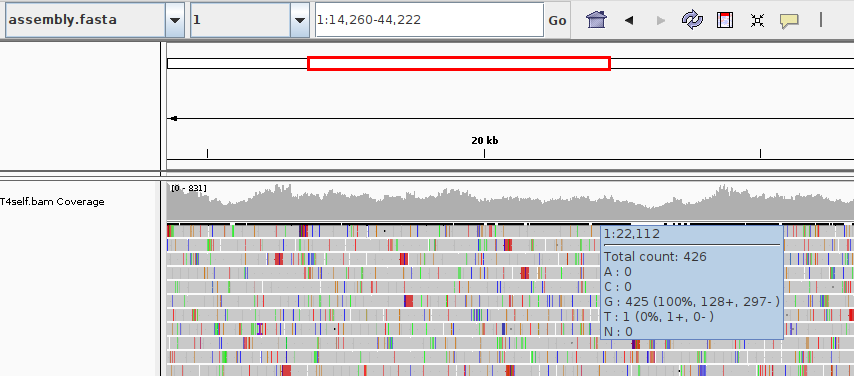
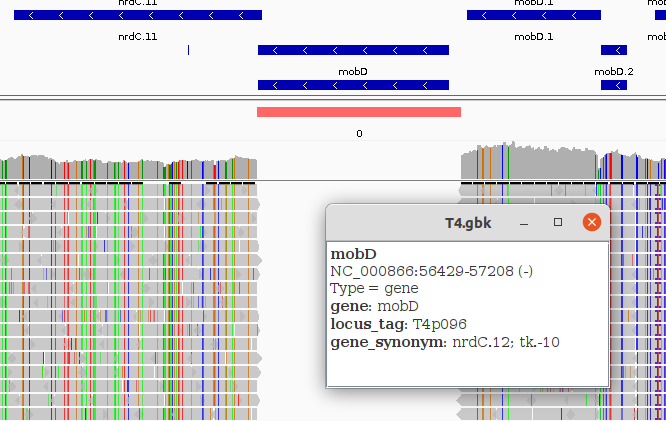
As you can see you will see each read as a gray bar, with colored stripes to highlight mismatches. IGV will automatically generate a coverage track in gray.
We can use bamToCov to convert a BAM file in a coverage BED file. I will use "head" to have a sneak peek at the output:
bamtocov aln/T4.bam | headThe output is in the format chromosome name, start, end, coverage:
1 0 2 193
1 2 3 203
1 3 4 206
1 4 5 207
1 5 6 210
1 6 7 213
1 7 8 234
1 8 9 247
1 9 11 249
1 11 12 250
🖊️ Do it yourself
Download the reference genome of Phage T4 and align the reads against the reference, instead of the assembly. The output will have some uncovered reagions like:
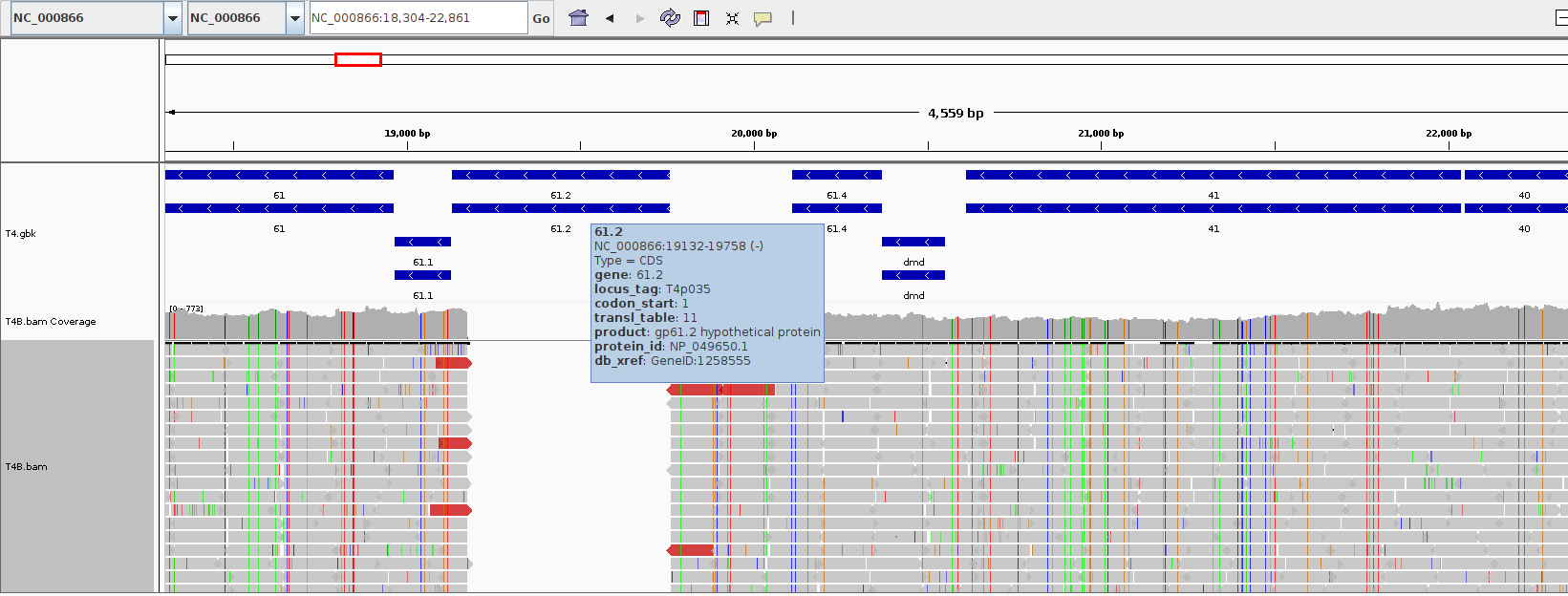
We are now interested in selecting those regions for a quick inspection.
Rendering alignments in IGV is a resource intensive task, so we can zoom out up to some kilobases and then the coverage and reads will disappear. This problem is not present with the more compact BED files so we can create a BED file with our "uncovered regions" and then use it as a guide to inspect the uncovered regions.
# Remember to use the BAM file generated against the reference, not the assembly!
bamtocov aln/T4-ref.bam | grep -w 0 > empty.bedFrom IGV load the reference genome (from "Genomes") and then some tracks (the BAM, the BED and optionally the annotation file) from "File".
As you can see the BED file will render also for the whole genome, while the BAM file won't.
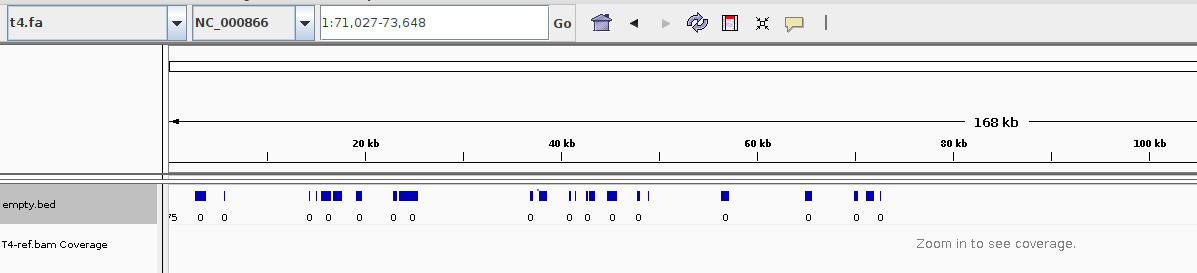
And if we zoom we can understand the reason for the missing parts