![]()

MAPpoly (v. 0.3.3) is an R package to construct genetic maps in diploids and autopolyploids with even ploidy levels. In its current version, MAPpoly can handle ploidy levels up to 8 when using hidden Markov models (HMM) and up to 12 when using the two-point simplification. When dealing with large numbers of markers (> 10,000), we strongly recommend using high-performance computing (HPC).
In its current version, MAPpoly can handle the following types of datasets:
- CSV files
- MAPpoly files
- Dosage based
- Probability based
- fitPoly files
- VCF files
MAPpoly also is capable of importing objects generated by the following R packages
The mapping strategy uses pairwise recombination fraction estimation as the first source of information to position allelic variants in specific homologs sequentially. The algorithm relies on the multilocus likelihood obtained through a hidden Markov model (HMM) for situations where pairwise analysis has limited power. The derivation of the HMM used in MAPpoly can be found in Mollinari and Garcia, 2019. The computation of the offspring's genotypes probabilities and haplotype reconstruction, as well as the preferential pairing profiles, is presented in Mollinari et al., 2020.
To install MAPpoly from the The Comprehensive R Archive Network (CRAN) use
install.packages("mappoly")You can install the development version from Git Hub. Within R, you need to install devtools:
install.packages("devtools")If you are using Windows, please install the the latest recommended version of Rtools.
To install MAPpoly from Git Hub use
devtools::install_github("mmollina/mappoly", dependencies=TRUE)For further QTL analysis, we recommend our QTLpoly package. QTLpoly performs random-effect multiple interval mapping (REMIM) in full-sib families of autopolyploid species based on restricted maximum likelihood (REML) estimation and score statistics, as described in Pereira et al. 2020.
We recently released VIEWpoly. VIEWpoly provides a graphical user interface to integrate, visualize and explore results from linkage and quantitative trait loci analysis, together with genomic information for autopolyploid (and diploid) species. The app is meant for interactive use and allows users to optionally upload different sources of information, including gene annotation and alignment files, enabling the exploitation and search for candidate genes in a genome browser. VIEWpoly supports inputs other than MAPpoly's, including polymapR, diaQTL, QTLpoly, and polyqtlR.

{kind=link}
{kind=link}
{kind=link}
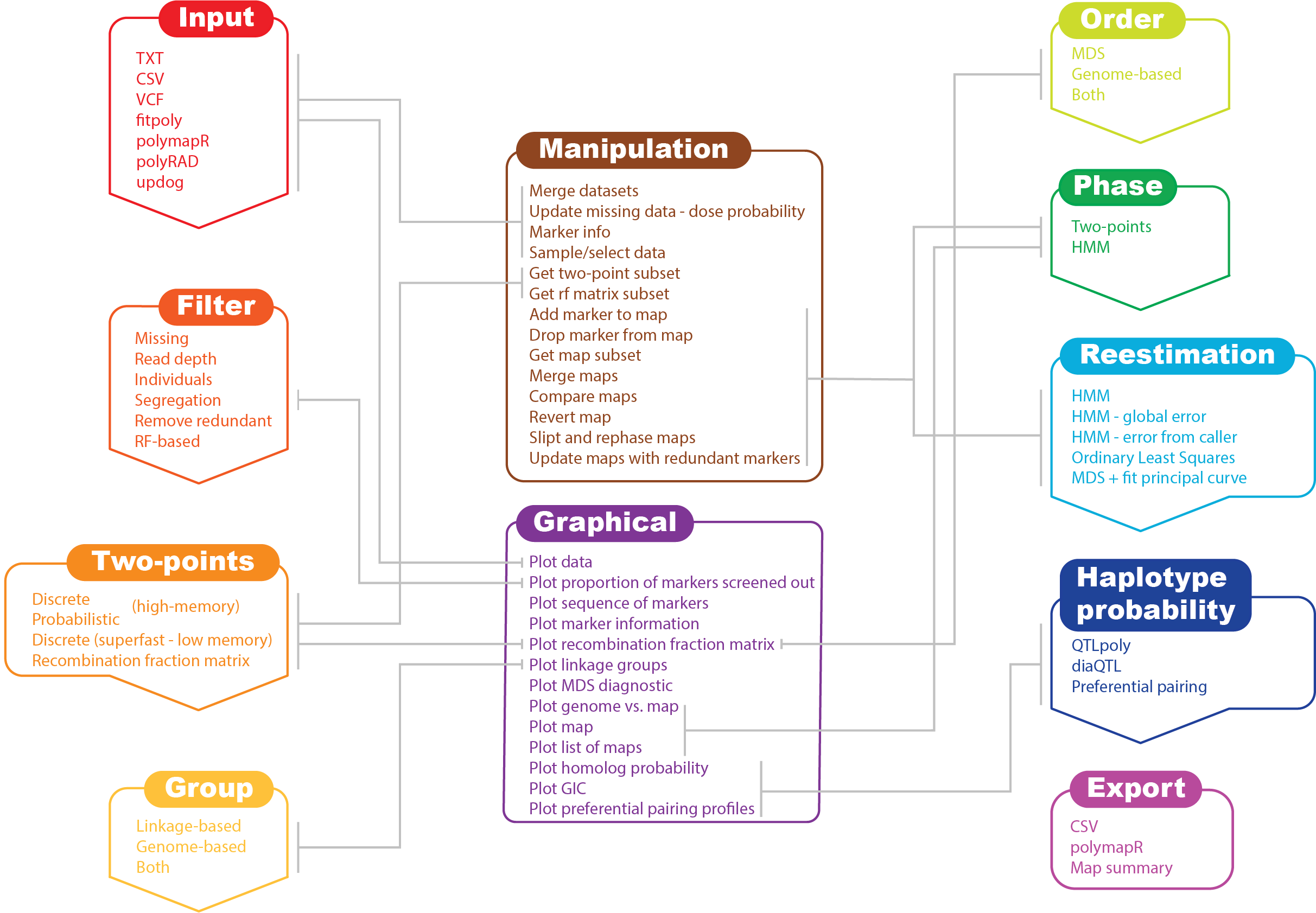
- To access the MAPpoly vignette from R, use
vignette("mappoly_startguide") - Building a genetic map in a tetraploid potato full-sib population using MAPpoly
- Building a genetic map in an hexaploid full-sib population using MAPpoly
- Real datasets
- Hexaploid sweetpotato VCF dataset (Beauregard x Tanzania) obtained using VCF2SM
- Tetraploid potato with dosage call in MAPpoly format
- Tetraploid potato with dosage call in CSV format
- Tetraploid potato with dosage probabilities in MAPpoly format
- Tetraploid potato in CSV format obtained using ClusterCall
- Compressed tetraploid potato with dosage probabilities obtained using fitPoly
- Simulated datasets
- Polyverse - the polyploid R universe (a Lindsay Clark's initiative)
# Enable this universe
options(repos = c(
polyploids = 'https://polyploids.r-universe.dev',
CRAN = 'https://cloud.r-project.org'))
# Install some packages
install.packages('mappoly')-
Variant Calling
-
Simulations
-
Genotype calling
- ClusterCall: Automated tetraploid genotype calling by hierarchical clustering
- fitPoly: Genotype Calling for Bi-Allelic Marker Assays
- polyRAD: Genotype Calling with Uncertainty from Sequencing Data in Polyploids and Diploids
- SuperMASSA: Graphical Bayesian inference tool for genotyping polyploids
- updog: Flexible Genotyping for Polyploids
- VCF2SM: Python script that integrates VCF files and SuperMASSA
-
Genetic mapping in polyploids
-
Haplotype reconstruction
-
QTL mapping
-
Visualization
- Rose Rosette Disease Resistance Loci Detected in Two Interconnected Tetraploid Garden Rose Populations (Lau et al., 2022)
- VIEWpoly: a visualization tool to integrate and explore results of polyploid genetic analysis. (Taniguti et al., 2022)
- Genetic Dissection of Early Blight Resistance in Tetraploid Potato. (Xue et al., 2022)
- Haplotype reconstruction in connected tetraploid F1 populations (Zheng et al., 2021)
- QTL mapping in outbred tetraploid (and diploid) diallel populations (Amadeu et al., 2021)
- Using probabilistic genotypes in linkage analysis of polyploids. (Liao et al., 2021)
- Discovery of a major QTL for root-knot nematode Meloidogyne incognita resistance in cultivated sweetpotato Ipomoea batatas. (Oloka, et al., 2021)
- Quantitative trait locus mapping for common scab resistance in a tetraploid potato full-sib population. (Pereira et al., 2021)
- The recombination landscape and multiple QTL mapping in a Solanum tuberosum cv.'Atlantic'-derived F1 population. (Pereira et al., 2021)
- High-Resolution Linkage Map and QTL Analyses of Fruit Firmness in Autotetraploid Blueberry (Cappai et al., 2020)
- When a phenotype is not the genotype: Implications of phenotype misclassification and pedigree errors in genomics-assisted breeding of sweetpotato Ipomoea batatas (L.) Lam.(Gemenet et al., 2020)
- Quantitative trait loci and differential gene expression analyses reveal the genetic basis for negatively associated beta-carotene and starch content in hexaploid sweetpotato [Ipomoea batatas (L.) Lam.] (Gemenet et al., 2020)
- Multiple QTL Mapping in Autopolyploids: A Random-Effect Model Approach with Application in a Hexaploid Sweetpotato Full-Sib Population. (Pereira et al., 2020)
This package has been developed as part of the Genomic Tools for Sweetpotato Improvement project (GT4SP) and SweetGAINS, both funded by Bill & Melinda Gates Foundation. Its continuous improvement is made possible by the project AFRI-Grant: A Genetics-Based Data Analysis System for Breeders in Polyploid Breeding Programs and SCRI-Grant: Tools for polyploids, funded by USDA NIFA.


NC State University promotes equal opportunity and prohibits discrimination and harassment based upon one’s age, color, disability, gender identity, genetic information, national origin, race, religion, sex (including pregnancy), sexual orientation and veteran status.