Before opening a new issue here, please check the appropriate help channel on the bioBakery Support Forum (https://forum.biobakery.org) and consider opening or commenting on a thread there.
Please direct to https://github.com/biobakery/halla_legacy for the 0.8.17 version of HAllA.
Given two high-dimensional 'omics datasets X and Y (continuous and/or categorical features) from the same n biosamples, HAllA (Hierarchical All-against-All Association Testing) discovers densely-associated blocks of features in the X vs. Y association matrix where:
- each block is defined as all associations between features in a subtree of X hierarchy and features in a subtree of Y hierarchy,
- and a block is densely associated if (1 - FNR)% of pairwise associations are FDR significant (FNR is the pre-defined expected false negative rate)
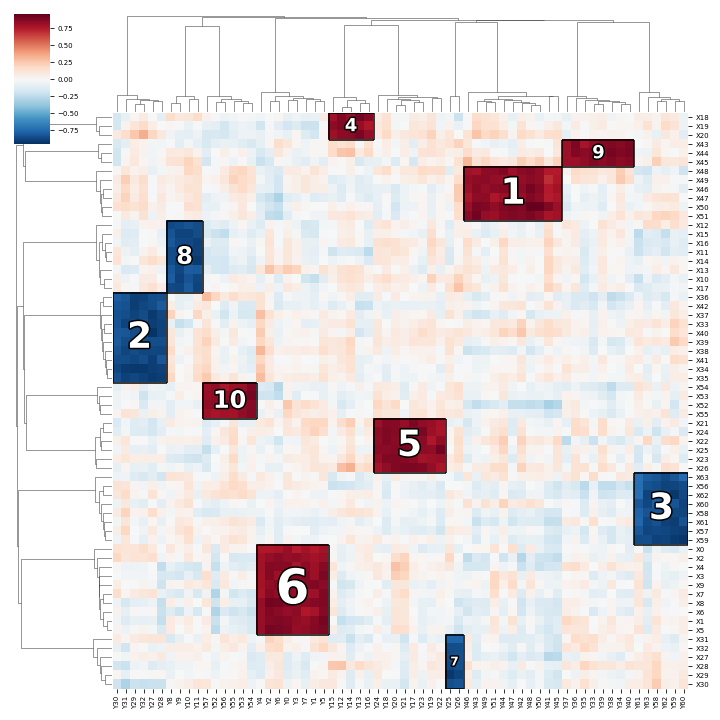
Example codes can be found under examples directory.
- Python>=3.8
- R
pip install halla
Note: Please install the following R packages manually:
git clone https://github.com/biobakery/halla.git
cd halla
python setup.py develop
Other than Python (version >= 3.7) and R (version >= 3.6.1), install all required libraries listed in requirements.txt, specifically:
Requirements Python packages:
- jenkspy (version >= 0.1.5)
- Matplotlib (version >= 3.5.3)
- NumPy (version >= 1.19.0)
- pandas (version >= 1.0.5)
- PyYAML (version >= 5.4)
- rpy2 (version >= 3.3.5) - Notes on installing
rpy2in macOS - scikit-learn (version >= 0.23.1)
- SciPy (version >= 1.5.1)
- seaborn (version >= 0.10.1)
- statsmodels (version >= 0.11.1)
- tqdm (>=4.50.2)
R packages:
# for MacOS - read the notes on installing rpy2:
# specifically run 'env CC=/usr/local/Cellar/gcc/X.x.x/bin/gcc-X pip install rpy2'
# where X.x.x is the gcc version on the machine **BEFORE** running the following command
conda install -c biobakery halla
Available pairwise distance metrics:
spearman(default for continuous data)pearsonmi(default for mixed/categorical data)nmixicordcor
There are three steps in HAllA:
The pairwise similarity matrix between all features in X and Y is computed with a specified similarity measure, such as Spearman correlation and normalized mutual information (NMI). This step then generates the p-value and q-value tables.
Note that for handling heterogeneous data, all continuous features are first discretized into bins using a specified binning method.
Hierarchical clustering on the features in each dataset is performed using the converted similarity measure used in step 1. It produces a tree for each dataset.
This recursive step is described in the pseudocode below:
def find_densely_associated_blocks(x, y):
x_features = all features in x
y_features = all features in y
if is_densely_associated(x_features, y_features):
report block and terminate
else:
# bifurcate one according to Gini impurities of the splits
x_branches, y_branches = bifurcate_one_of(x, y)
if both x and y are leaves:
terminate
for each x_branch and y_branch in x_branches and y_branches:
find_densely_associated_blocks(x_branch, y_branch)
initial function call: find_densely_associated_blocks(X_root, Y_root)
For example, given two datasets of X (features: X1, X2, X3, X4, X5) and Y (features: Y1, Y2, Y3, Y4) both hierarchically clustered in X tree and Y tree, the algorithm first evaluates the roots of both trees and checks if the block consisting of all features of X and Y are densely-associated (if %significance (%reject) >= (1 - FNR)%).

If the block is not densely-associated, the algorithm would bifurcate one of the trees. It would pick one of:
- [X1 X2][X3 X4 X5] >< [Y1 Y2 Y3 Y4] or
- [X1 X2 X3 X4 X5] >< [Y1 Y2 Y3][Y4]
based on the Gini impurity of the splits (pick the split that produces a lower weighted Gini impurity),

Once it picks the split with the lower impurity (let's say the first split), it will iteratively evaluate the branches:
- find densely-associated blocks in [X1 X2] vs [Y1 Y2 Y3 Y4], and
- find densely-associated blocks in [X3 X4 X5] vs [Y1 Y2 Y3 Y4]
and keep going until it terminates.
Thanks go to these wonderful people: