Using the DLTRS model
A tutorial for DLTRS (also known as Deleterious).
DLTRS [=the model] is a phylogenetic model for the evolution of a guest tree (gene family) inside a host tree (species tree). It models duplication, loss, lateral transfer (LGT) events, and sequence evolution with a relaxed molecular clock. It is strongly related to its sibling model, [DLRS DLRS], but extends the latter by incorporating support for transfer events.
The evolution of the guest tree G over the host tree S can be envisioned as occurring in separate phases:
- G is created by evolving down S by means of duplication, loss and transfer events according to a birth-death-like process. Lineages branch deterministically at speciations. In case of an LGT, a receiving species lineage contemporary with (but excluding) the donor species lineage is uniformly selected.
- Using the clock-like times of G, relaxed branch lengths are obtained by multiplying each branch timespan with an iid substitution rate according to a suitable distribution.
- Using G and its branch lengths, sequence evolution occurs over the tree according to a substitution model of choice to produce guest family sequences. Possibly, site rate variation occurs according to a gamma distribution.
Hence the name DLTRS = duplications, losses, transfers, rates & sequence evolution.
JPrIME-DLTRS [=the software application], colloquially known as Deleterious, is the Java application corresponding to the DLTRS model, and is used for inferring an unknown guest tree in light of a known and dated host tree and known sequence data for the leaves. The method relies on Bayesian inference using an MCMC framework. Thus, it will yield a posterior distribution of guest tree topologies and remaining parameters. Note, however, that in comparison with its sibling tool, [DLRS Delirious], there are some fairly significant differences in how the underlying probabilities are computed, and Deleterious is more computationally demanding than Delirious. Thus, if lateral gene transfer are highly improbable, Delirious will be a better choice.
Input:
- a file with a dated host tree.
- a file with a multiple sequence alignment (MSA) of guest tree leaf sequences.
- a file with a simple map relating the guest tree leaves to host tree leaves.
Output:
- a file with samples drawn from the posterior distribution.
- a supplementary info file with settings, proposal acceptance ratios, etc.
Since so many properties of Deleterious are shared with Delirious, we will often refer to the DLRS tutorial of the latter in order to keep information comprehensive and up-to-date. If you want to get started quickly, we suggest that you download and try Example 1 (below), which can be found below, and have a look at it while reading through the other parts of the guide.
DLTRS is not distributed as a binary, but is included in the JPrIME JAR file, which is downloadable.
The best reference for the DLTRS software is currently
- Sjostrand J, Tofigh A, Daubin V, Arvestad L, Sennblad B, Lagergren J (2014) A Bayesian method for analyzing lateral gene transfer. Syst Biol 63(3), 409-420
A typical workflow may look like this. More details are found below.
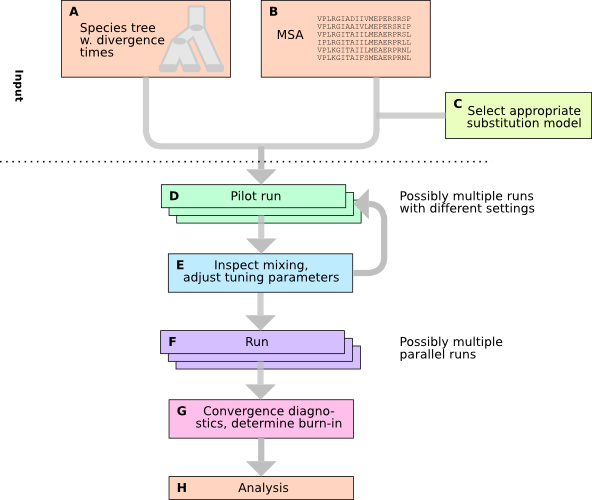
The required input files to Deleterious are exactly the same as for Delirious. Therefore, please consult the DLRS tutorial for details. As mentioned above, these consist of 1) a host tree with divergence times on the Newick format, 2) a multiple sequence alignment on the FASTA format, and 3) a file relating each sequence to each extant taxon.
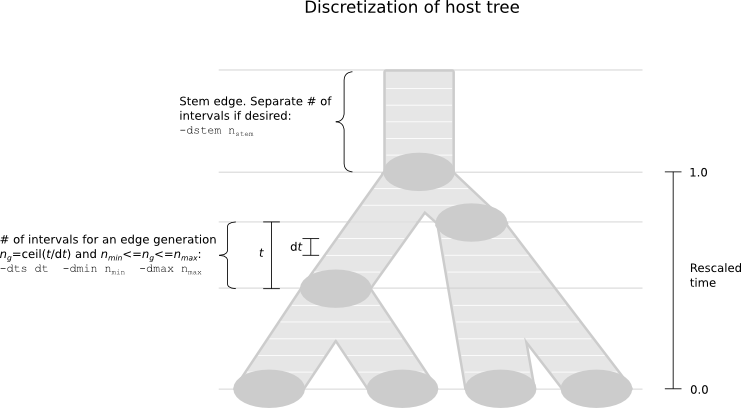
There is, however, a difference in the way that the host tree is discretized. As with Delirious, the host tree must be clock-like (ultrametric), with time 0 at the leaves and time >0 at the root, and a "stem" with timespan >0. The scale of the dates can be any unit: they will be rescaled internally prior to running so that the root time is 1.0, and the output will refer to this scaling, and not the original values. The scale factor is included in the info output file. The algorithm will use a discretization of the host tree based on sub-dividing contemporary "edge generations". The default settings may be changed according to the options shown below.
Output is virtually identical to Delirious output, see OUTPUT (below). The only major difference is the inclusion of an additional column for the LGT rate. The accompanying "info file" should be inspected for determining that MCMC mixing is performing adequately.
Deleterious is started as a command-line Java application. It is possible that future versions will come with a graphical user interface, a command-line based environment for managing options, or similarly. However, at the moment, you would start Deleterious by running e.g.:
java -jar jprime-X.Y.Z.jar Deleterious [options] <host tree> <msa> <leaf map>
where the JAR file of course refer to your current setup. See also the section on options below. We recommend using Oracle's HotSpot Java Virtual Machines due to their excellent performance, in which case a recommended heap size of 512 MB and a maximum heap size of 1024 MB would be specified with:
java -Xms512m -Xmx1024m -jar jprime-X.Y.Z.jar Deleterious [options] <host tree> <msa> <leaf map>
Note that these options refer to Java's VM itself, and are not options of Deleterious.
As in most MCMC applications, you typically conduct:
- pilot runs to assess acceptance ratios, mixing, and running time,
- if required, change tuning parameters to improve mixing,
- perform the actual runs, often starting 2-4 parallel chains.
We refer to the RUNNING section on running Delirious for more information.
Analysis is carried out in the same way as with Delirious; please see the corresponding DLRS ANALYSIS section for more details. MCMC chain output consists of a tab-delimited file which can be read with e.g. CRAN R, Matlab, or Octave. Note that under typical settings, Deleterious will output one column more than Delirious, namely the LGT rate. Trees are output as sorted Newick strings, and can easily be counted, analyzed for common clades etc. using external software (or even a shell). As always, one should discard an initial portion of the output as burn-in to make sure analyzed samples have converged to the posterior distribution. You can use e.g. VMCMC for testing MCMC chains for suitable burn-in cut-offs, acquire summary statistics, etc.
Delirious comes with a large number of user options; please don't feel overwhelmed! Some of the more commonly used ones are detailed below. You can always type
java -jar jprime-X.Y.Z.jar Deleterious -h
to get a more up-to-date description of all available options.
General options
-
-o <string>Output file for MCMC chain. The auxiliary info file will be appended with .info. -
-i <int>Number of iterations, e.g. 1000000. -
-t <int>Governs how often samples are taken, e.g. 200. -
-loutTo include a Newick tree with branch lengths among the samples and not only the plain Newick tree.
Substitution model
Deleterious can work with DNA, peptide and codon MSAs. As of yet, working with several independent loci is not supported.
-
-sm <string>The substitution model. Some common empirical models are included. However, you may also provide your own time-reversible model by inputting its stationary frequencies and its exchangeability matrix (not the transition matrix). -
-srcats <int>Number of discrete site rate categories for modeling gamma site rate heterogeneity. The default is 1 category. Note: Running time may in many cases grow linearly with the number of categories. Invariant sites are not modeled. -
-erpd <string>Probability distribution used for modeling iid rates of relaxed molecular clock. The default is the gamma distribution.
Initial values and leaving parameters out
-
-g <string>Initial guest tree. You can choose to create a random tree, a NJ tree based on sequence identity, or inputting your own tree, possibly with branch lengths. Recently, support for inferring a more sophisticated NJ tree was added with option-fp. Note: The guest tree must be rooted and bifurcating. -
-gfixFix the guest tree. -
-lfixFix the branch lengths. -
-dup <float>Duplication rate. You may fix this value by appending it with FIXED. -
-loss <float>Loss rate. Fix as above. -
-trans <float>Transfer rate. Fix as above. -
-erpdm <float>Relaxed molecular clock mean. Fix as above. -
-erpdcv <float>Relaxed molecular clock coefficient of variation. Fix as above. -
-srshape <float>Gamma site rate shape parameter. Fix as above.
Tuning parameters
In order to get good mixing, it might be necessary to tweak the tuning parameters. These come in several flavours, of which the most important ones are for 1) setting the audacity of the proposed new states, and 2) setting how often a state parameter should be perturbed.
-
-tng<param> <[float,float]>General pattern for tuning parameters of floating point state parameters. The specified values controls the proposal distribution's CV for the first and last iterations respectively. Try striving for acceptance ratios of around 0.2-0.5. To see whether this is the case, inspect the info files of pilot runs. If acceptance ratio too low, decrease the CV from its default, if too high, increase the CV. -
-tngw<param> <[float,float]>General pattern for setting the weights for how often parameters are perturbed with respect to each other.
Discretization
Should the default discretization settings not be satisfactory, you can change them. A denser discretization means higher precision but also higher running time. See the figure above for a more detailed description.
-
-dts <float>Approximate timestep between discretization points in an edge generation. Remember that this refers to the rescaled species tree where the root-to-leaf time is 1. -
-dmin <int>The minimum number of discretization slices of an edge generation. -
-dmax <int>The maximum number of discretization slices of an edge generation. -
-dstem <int>The number of discretization slices of the stem edge.
A microbial gene family of Mollicutes within the Firmicutes phylum. These typically lack a cell wall, and are often pathogenic, such as Mycoplasma.
- dltrs_example_1.stree (host tree)
- dltrs_example_1.fa (protein MSA)
- dltrs_example_1.map (guest-to-host leaf map)
Fancying 1,000,000 iterations, sampling every 200th iteration, and using the JTT substitution model, we may get something like:
java -Xms128m -Xmx256m -jar jprime-X.Y.Z.jar Deleterious -o dltrs_example_1.mcmc -i 1000000 -t 200 -sm JTT dltrs_example_1.stree dltrs_example_1.fa dltrs_example_1.map
Another Mollicutes gene family. This family has a weak signal, resulting in many low-probability trees. Here, a consensus tree may provide some information. Future extensions of JPrIME and related tools may aid in inspecting the tree space in other fashions.
- dltrs_example_2.stree (host tree)
- dltrs_example_2.fa (protein MSA)
- dltrs_example_2.map (guest-to-host leaf map)
Here, we might try to improve the MCMC mixing a bit by reducing the duplication and loss rate suggestion boldness, and increasing the relative proportion of branch-swapping operations:
java -Xms128m -Xmx256m -jar mypath/jprime-X.Y.Z.jar Deleterious -o dltrs_example_2.mcmc -i 1000000 -t 200 -sm JTT -tngdup [0.3,0.3] -tngloss [0.3,0.3] -tngwg [4,4] dltrs_example_2.stree dltrs_example_2.fa dltrs_example_2.map
Since Deleterious shares most aspects with its sibling application Delirious, we refer to the [DLRS#FAQ FAQ of Delirious] for most issues. There are, however some differences, notably in discretization and running time.
Q1) What is a reasonable discretization for Deleterious? And what about tree sizes?
A1) That depends. Deleterious is an advanced and already fairly optimized application, which solves systems of ordinary differential equations and performs sophisticated dynamic programming during MCMC iterations. As such, it is time-consuming, and you will have to strike a balance between speed and precision in terms of discretization density. Note, however, that the discretization is carried out on an edge generation-basis (see the figure above). Therefore, an edge generation typically needs fewer points than an edge in Delirious. Also, since transfer events are supported, Deleterious does not need as many points as Delirious on the stem edge in order to cope with odd trees during tree exploration. Try using the default discretization, and if it is too demanding, decrease the density gradually. You cannot, however, expect to run Deleterious on host trees larger than, say, 20 extant taxa. In that case wou will have to resort to more crude methods. Note also that the running time is linear in the number of guest tree sequences.
Q2) LGT events always occur between disjoint donor- and recipient species lineages. What about intra-species transfer?
A2) This is an interesting question. Such events will appear as duplications in the model. Therefore, the inferred duplication rate will encompass both intra-species transfers and, e.g., tandem duplications, whereas the transfer rate will capture inter-species transfer.