Pvalue.py is new and compute Pvalue for the Alpha and Beta (See below)
BitEpi is software for an exhaustive search of higher-order epistatic interactions between genomic variants (SNPs) given a binary phenotype (Case/Control).
It can search for 2-SNP (pairwise) interactions as well as 3-SNP and 4-SNP (Higher-Order) interactions. It also processes SNPs individually (1-SNP test)
BitEpi performs an exhaustive search that means it tests all possible combinations of SNPs. For example, the 4-SNP search of 50 SNPs (data.csv) requires to test all "4 out of 50" 4-SNP combinations (230300 4-SNP combinations).
BitEpi implements efficient multi-threading (parallelization) such that each thread tests almost the same number of combinations.
BitEpi could perform 2 different association analysis: Beta and Alpha
Beta analysis measures the association power of an SNP (1-SNP), a pair of SNPs (2-SNP), a combination of three SNPs (3-SNP) and a combination of four SNPs (4-SNP). Beta represents the combined association power. Beta is computed as the weighted average purity (Gini-Index) of each row of the contingency table where the weight is the fraction of samples in that row. The below example shows how we compute Beta for an individual SNPs (1-SNP Beta).
| Genotype | # Cases | # Controls | Purity | Weight | Purity * Weight |
|---|---|---|---|---|---|
| 0/0 | 5 | 10 | ((5*5)+(10*10))/(15*15) = 0.555 | 15/30 = 0.5 | 0.278 |
| 0/1 | 2 | 4 | ((2*2)+(4*4))/(6*6) = 0.555 | 6/30 = 0.2 | 0.111 |
| 1/1 | 8 | 1 | ((8*8)+(1*1))/(9*9) = 0.802 | 9/30 = 0.3 | 0.241 |
| Beta (sum): | 0.630 |
2-SNP, 3-SNP, and 4-SNP Beta are computed in the same way. the only difference is that there are more rows in the contingency table (9, 27 and 81 rows respectively).
Alpha analysis measure the interactions effect size between SNPs. In other words, Alpha is the gain in the association power due to interaction between SNPs. Alpha computed as below where B_ indicates Beta value.
- (4-SNP) Alpha WXYZ = B_WXYZ - Max(B_WXY, B_WXZ, B_WYZ, B_XYZ)
- (3-SNP) Alpha XYZ = B_XYZ - Max(B_XY, B_XZ, B_YZ)
- (2-SNP) Alpha XY = B_XY - Max(B_X, B_Y)
- (1-SNP) Alpha X = B_X - B_0
B_0 is the purity (Gini-Index) of the samples in the dataset (CONSTANT). Given r case and q controls, B_0 is computed as (r^2 + q^2) / (r+q)^2. 1-SNP Alpha is similar to 1-SNP Beta with an offset (B_0). While the minimum value for Beta depends on the dataset characteristics (B_0), the minimum value for Alpha is always 0.
High value for Beta does not necessarily indicate strong interactions.
For example, let's say
- Beta A = 0.90 (association power of SNP A)
- Beta B = 0.03 (association power of SNP B)
- Beta AB = 0.92 (association power of pair of A and B)
In this example, the Beta AB is high. However, it is driven mainly by Beta A but not the interaction between A and B. Another example is
- Beta X = 0.51
- Beta Y = 0.53
- Beta XY = 0.75
In this example, Beta XY is not as high as Beta AB. However, it is not driven by individual SNPs but by the strong interaction between X and Y.
The Alpha analysis reveals this:
- Alpha AB = Beta AB - Max( Beta A, Beta B) = 0.92 - Max(0.90, 0.03) = 0.02 (Low Alpha -> no interaction)
- Alpha XY = Beta XY - Max( Beta X, Beta Y) = 0.75 - Max(0.51, 0.53) = 0.22 (High Alpha -> strong interaction)
best Mode
If you run BitEpi in the best mode, BitEpi finds the best 2-SNP, 3-SNP and 4-SNP interactions for each SNP (maximize Alpha) and then report Beta and Alpha for each SNPs and its best 2-SNP, 3-SNP and 4-SNP interactions.
Notes
- To computer (N)-SNP Alpha the program needs (N-1)-SNP Beta. To avoid computational redundancy, BitEpi computes all (N-1)-SNP Beta and store them (n-1) dimension array. For 4-SNP search, the size of this array is (v^3)*8 bytes where v is the number of SNPs in the dataset (i.e for 4000 SNPs 256 GB of Memory). This is a bottleneck of the program. Using 3 dimension array to store 3-SNP combination results in memory redundancy. However, these data are accessed so frequently and the array leads to the fastest access.
- You can combine different analyses, for example, you can do 3-SNP Beta and 2-SNP alpha in the same run.
- You can choose to report all SNP combinations that exceed specific Alpha and Beta threshold. However, there are no guidelines on how to choose such a threshold. It would be easier if you ask for the top N SNP combinations (with the highest Alpha or Beta) to be reported. This option is implemented in BitEpi with minor computational cost (see command line parameter Notes)
- If you parallelize the program on so many threads such that each thread process a very small number of combinations to test (less than MIN_COMB_IN_JOB in the code). The program exit without performing the job. In this case, you should use a smaller number of threads.
BitEpi is Fast
BitEpi uses fast bitwise operations to count samples with a different genotype. That is why we call it BitEpi. BOOST (pairwise search only) and MPI3SNP (3-SNP search only) also use bitwise operations for this purpose. However, the bitwise method used in BitEpi is different and better suits 3-SNP and 4-SNP (higher-order) analysis. As a consequence, BitEpi is slower than BOOST for 2-SNP search but faster than anything else for 3-SNP and 4-SNP search (to the best of our knowledge).
Python Interface
BitEpi Python Interface is implemented in a separate GitHub page (bitepi-python). You can find examples and descriptions there. It may take time for changes on this repository to be reflected on the bitepi-python repository.
Command-Line Interface (CLI) Arguments
| Argument | Description |
|---|---|
| -i [str] | Path to input file (CSV or plink bfile). |
| Default is CSV (see -bfile) | |
| -bfile | If present, -i argument is considered as |
| plink bfile prefix (_.bed, _.bim, *.fam) | |
| -o [str] | Output prefix |
| -bfile | Read input file as PLINK 1.9 .bed format |
| -sort | Sort the output |
| -best | Find the best interactions for each SNP |
| -t [int] | Number of threads |
| -b1 [thr] | Perform 1-SNP Beta analysis |
| -b2 [thr] | Perform 2-SNP Beta analysis |
| -b3 [thr] | Perform 3-SNP Beta analysis |
| -b4 [thr] | Perform 4-SNP Beta analysis |
| -a1 [thr] | Perform 1-SNP Alpha analysis |
| -a2 [thr] | Perform 2-SNP Alpha analysis |
| -a3 [thr] | Perform 3-SNP Alpha analysis |
| -a4 [thr] | Perform 4-SNP Alpha analysis |
notes
- If thr<1 then thr is the minimum threshold on alpha and beta to be reported in the output.
- If thr>=1 then thr is the number of top hits in alpha and beta to be reported in the output.
- If thr>=1 and more than 1 thread are used, then each thread reports thr top hits and the output files are merged. So (t*thr) top hits will be reported. You can use -sort option and only consider the top thr record.
- thr is optional. If you don't pass a thr the program computes the metric but it does not report anything (performance testing).
- If all interactions should be reported set thr to 0.
CSV Input Format
- The first row includes labels: 1 and 0 for cases and controls respectively
- The first column includes SNP unique ids. BitEpi does not check the uniqueness.
- The first entry (first col and first row) is ignored.
- All other entry represents genotype and can be 0, 1 or 2 for 0/0, 0/1 and 1/1 genotype (phased genotype are not supported).
Output File Naming
- output_prefix.best.csv
- output_prefix.Alpha.Order.csv
- output_prefix.Beta.Order.csv
- output_prefix.Alpha.Order.FirstJobIndex.LastJobIndex.csv
- output_prefix.Beta.Order.FirstJobIndex.LastJobIndex.csv
Use the g++ commands in compile.sh or just run it
$ cd BitEpi
$ bash compile.shSee examples in runme.sh or just run it
$ cd BitEpi
$ bash runme.shGAMETES is an epistasis simulator (Paper) used in several publications to measure the accuracy of different methods. GAMETES_2_EPI.sh converts the GAMETES output to the BitEpi input format and the plink transposed format (tplink). You can process tplink file with MIP3SNP and BOOST (implemented in plink) for performance testing. Note that MDR could process GAMETES output directly. An example GAMETES output is provided: GAMETES_Example_Output.txt
See below example
$ bash GAMETES_2_EPI.sh sampleData/GAMETES_Example_Output
$ ls sampleData/GAMETES_Example_Output*VCF_2_EPI.sh is a script that uses bcftools and Linux commands to convert a VCF file into BitEpi format. Since VCF file does not include phenotype, the binary phenotype is read from a tab-separated (tsv)sample annotation file (1 line per sample with a header line). You should specify which column holds the binary phenotype ("True" or "False"). The script prints important points about the input argument you should consider. We include an example VCF file (based on 1000-Genome project) and a sample annotation file (with a synthetic phenotype) to try the script. Four important SNPs used to simulate phenotype are listed in Hipster.known.txt.
See below example
$ bash VCF_2_EPI.sh sampleData/Hipster.vcf.bgz sampleData/Hipster.tsv 2
$ less -S sampleData/Hipster.vcf.bgz.epi.csv2-SNP (pairwise) and 3-SNP (triplet)
- Models
- Datasets and Models
- GAMETES format only (use the convertor above)
2-SNP data simulated in MACOED including ME (Marginal Effect) and NME (None Marginal Effect)
- Data
- Models are not provided.
- Data can be also downloaded from (its in csv not tsv) here
- GAMETES format only (use the converter above)
Performance (runtime) testing datasets:
- Data
- Available in GAMETES, BitEpi and transposed plink formats.
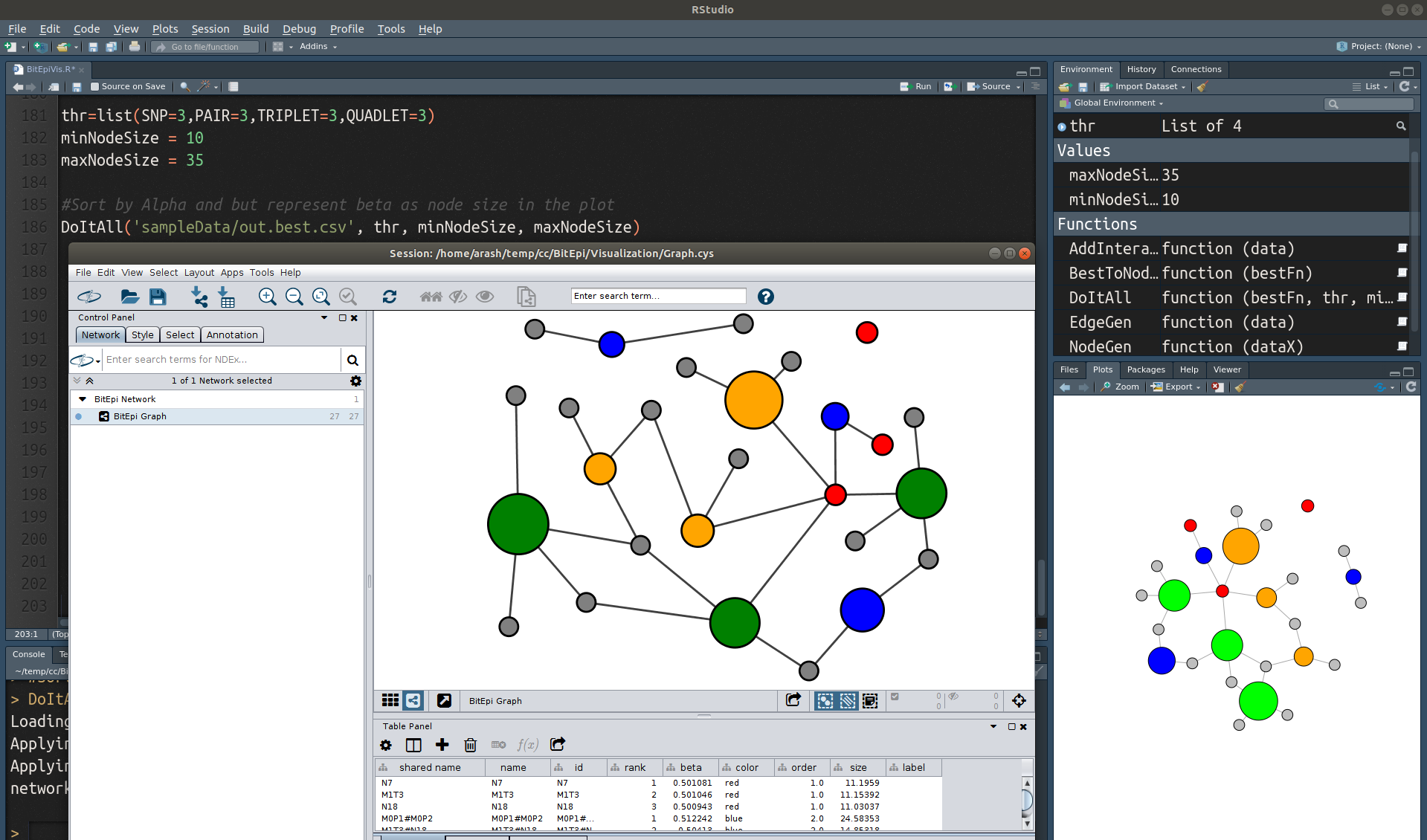
You can Visualize the best interactions (see best mode described above) using a Cytoscape Graph. BitEpiVis.R provides a function that reads the output of BitEpi "-best" analysis and turn it to an interactive Cytoscape graph. It also generates a static igraph plot in Rstudio. (See the example screenshot below). In the graph, you can select and move nodes around. Node color represents if the node is an SNP node or an interaction node (see below). Node size represents the combined association power (Beta).
Initialization
To get a nice plot in Cytoscope you should first import our style file BitEpiCytoscapeStyle.xml. Open Cytoscape and from File menu select Import and then select Style from File... and choose BitEpiCytoscapeStyle.xml. In the Style tab of the Control Panel, select BitEpi in the style dropdown menu. In the Style tab click on the menu button (three horizontal lines) and select Make Current Style Default
Run the R code
As you can see in the example screenshot, at the end of the BitEpiVis.R the thr variable is defined. It is the number of most significant SNPs (1-SNP) and interactions (2-SNP, 3-SNP, and 4-SNP) you want to include in the graph (based on Alpha).
The DoItAll function takes the path to the BitEpi best output and generates the graph. You should leave Cytoscape open when you run the R code. Once the Graph appears in Cytoscape, from Layout menu chooses Grid Layout (make sure BitEpi style is selected before that). Now you can see the nice plot.
You can drag and drop nodes to create your custom layout. When you click on a node and select it, it turns to yellow and you can see its details in the table below the graph. See the example screenshot.
Node Colors
Interactions are shown with Blue, Orange, and Green for 2-SNP, 3-SNP and 4-SNP significant interactions you choose to see. Significant SNPs are shown in Red. Other SNPs are shown in grey (they are there because they are part of one of the significant interaction). You can change the colors using the Color variable defined at the top of the R code.
Node Sizes
You cannot compare the node size of two different plot as node sizes are scaled to make all nodes in each plot visible. You can change the node size scale using minNodeSize and maxNodeSize in the R code (see example screenshots). Note that grey nodes represent the minimum node size.
Color=list(SNP='red',PAIR='blue',TRIPLET='orange',QUADLET='green', OTHER='gray')
thr=list(SNP=3,PAIR=3,TRIPLET=3,QUADLET=3)
minNodeSize = 10
maxNodeSize = 35
DoItAll('sampleData/out.best.csv', thr, minNodeSize, maxNodeSize)
Given genotype dataset in plink bfile format and list of interactions identified by BitEpi, Pvalue.py computes the Alpha and Beta Pvalue for each interaction (combination of SNPs) in the list of interactions.
For every SNPs combination, we randomize the phenotype (response variable) many times. We compute alpha and beta for each permutation. Then we find the best distribution that fit the data. we test the following four scipy distributions: gamma, norm, lognorm, expon. Given the best distribution we computed one-tailed Pvalue (1-cdf) for alpha and beta. Note that different distribution is fitted for alpha and beta.
Note that this python code is slow and is not optimized for performance.
It also implements a special mode where it randomly combines SNPs (assume they interact) and then it computes the Alpha and Beta and well as the corresponding Pvalue for them.
The command line usage examples are as follow:
python3 Pvalue.py rnd sampleData/bfile 5 1000 sampleData/pval-rnd.tsv 2
python3 Pvalue.py epi sampleData/bfile 5 1000 sampleData/pval-epi.tsv sampleData/bfile.Alpha.2.csv
in the first command
- "rnd" means to combine SNPs randomly and not from list of interactions
- "sampleData/bfile" is the path+prefix for plink bfile
- "5" is the number of SNP-combinations to process
- "1000" is the number of time random phenotype generated for each SNP-combination.
- "sampleData/pval-rnd.tsv" is the output file
- "2" is the number of SNPs in each SNP-combination
the second command is similar to the first command except
- "epi" means to read the list of interaction (Alpha or Beta output file from BitEpi)
- "5" means to process only the top 5 interactions in the list
- "sampleData/bfile.Alpha.2.csv" is the list of interactions (BitEpi output file)
The output file contains the following columns:
- FirstCol: The Alpha or Beta value computed by BitEpi (0 in rnd mode)
- SNPs: List of SNPs in the SNP-Combination
- beta: beta value computed by Pvalue.py
- alpha: alpha value computed by Pvalue.py
- alpha-dist: name of distribution fited to the alpha values
- alpha-distPval: Pvalue for alpha distribution fitness
- alpha-pval: Pvalue for the alpha based on the distribution
- beta-dist: name of distribution fited to the beta values
- beta-distPval: Pvalue for beta distribution fitness
- beta-pval: Pvalue for the beta based on the distribution
BitEpi is published in bioRxiv and is submitted to Bioinformatics (under review). For citation see cite.bib in Publication folder.