![]()


- It finds novel enriched pathways between two classes in comparison
- It exploits the novel paradigm of patient similarity networks to detect pathways as no other method
- It generates information and data to further manual analysis and explorations
- It can be applied of multiple omics
- It does not require technical parameters, only the patient's/samples's information
- It allows to plot an enriched pathway in form of patient similarity network to have a beautiful informative image for the paper which allows to highlight the similarities and dissimilarities between patients
- It performs an outlier dection and retrieves how much each patient is likely to not fit in its own class defined by its clinical information
- It allows to explore the signature pathway of a patient class with a graphical interface
Giudice Luca, et al., Simpati: pathway-based classifier exploits patient similarity networks for cancer stage prediction
Simpati is available on R, you can install it by:
devtools::install_github(
repo="LucaGiudice/Simpati",
ref = "main",
dependencies = "Depends",
upgrade = "always",
quiet = F
)There are the following vignettes:
- A Quick Start of Using Simpati Package and introduction to the results
- Advanced workflow of Simpati performed with all the operations explicit
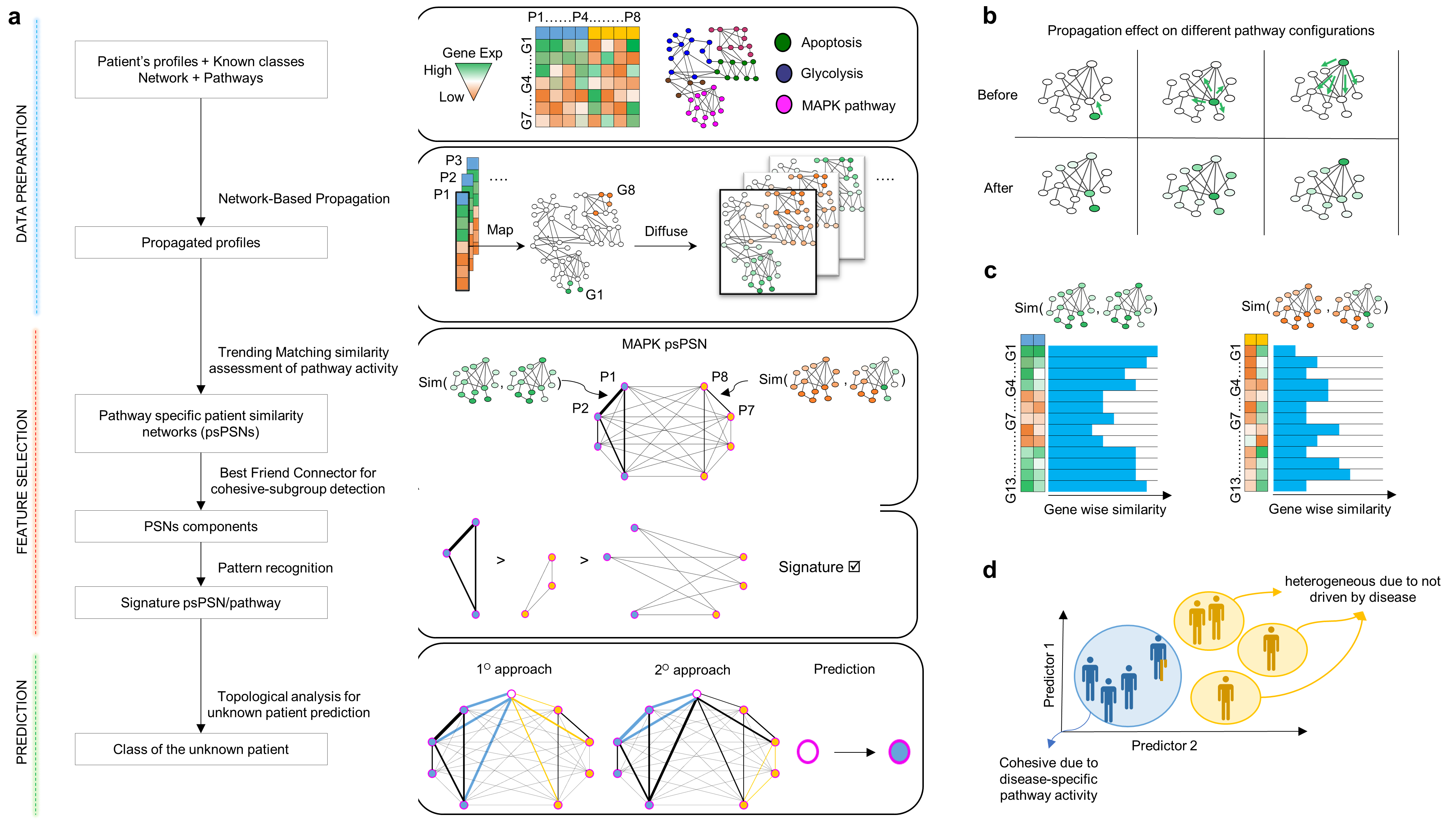
The cores of Simpati workflow:
Frame A shows the workflow
- Patient profiles colored by the class of belonging are described by biological features as genes. A gene-gene interaction network together with pathways are further input data required by the software.
- All profiles are individually propagated over the network. The profile’s values are replaced by scores that reflect the gene’s starting information and connections.
- Simpati proceeds by creating a patient similarity network for each pathway (psPSN). The pairwise similarity evaluates how much two patients have a similar pathway activity.
- The psPSN is decomposed into three components. Two with the intra-similarities of the class specific representative patients, while one with the inter-similarities between them. If the similarities of one class dominate over the other two components, then the psPSN is signature.
- The latter is ultimately used to classify. An unknown patient is classified based on how much is like the other patients and on how much fits in the class specific representatives.
Frame B shows the effect of the propagation
- The position of the gene and its level of expression leads a patient to act on the other genes and so on a pathway in a unique way.
Frame C shows the patient similarity used in Simpati
- It evaluates how much the genes between two patients are close and high in term of propagation values. Two patients that act on a pathway from the same gene positions and with the same expression values get the maximum similarity.
Frame D illustrates the biological interpretation behind a signature psPSN.
- One class is cohesive because the disease is leading the patients to alter similarly the same pathway. One class is sparse because led by multiple factors.
Few lines of code to perfrom Simpati analysis:
library(Simpati)
#Get omic-specific patient profiles and their clinical data
geno=tcga_data$LIHC$`LIHC_Mutation-20160128`$assay_df;see(geno)
info=tcga_data$LIHC$`LIHC_Mutation-20160128`$clin_df;see(info)
info=info[,c("patientID","pathologic_stage")]
#Set name of the dataset
dataset_name="LIHC"
#Set the semantic type of the disease for the disgnet enrichment
disease_type=tcga_data$LIHC$semantic_type
#Set key words associated to the patient's disease
key_words=tcga_data$LIHC$key_words
#Gene interaction network
net=huri_net_l$net_adj
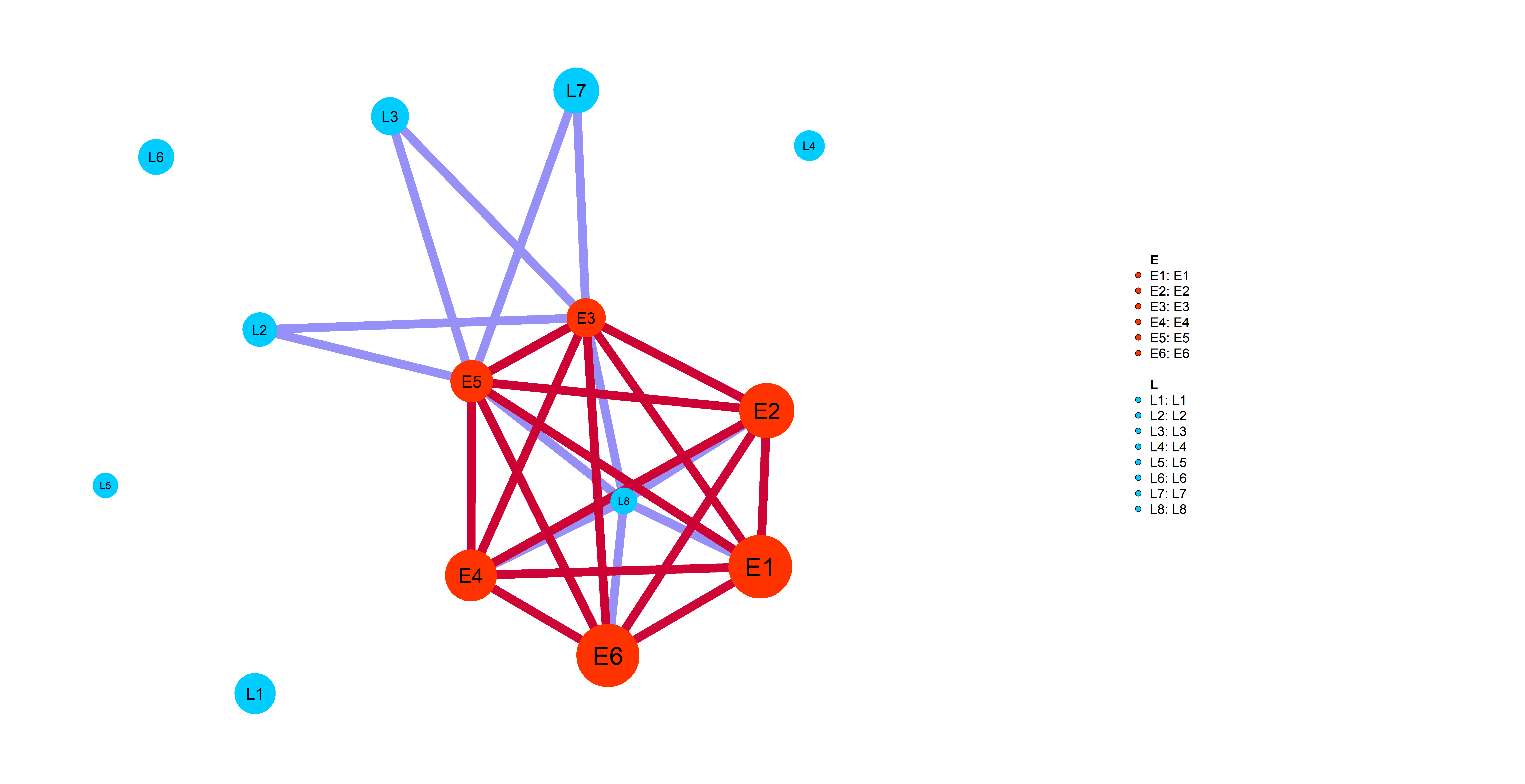
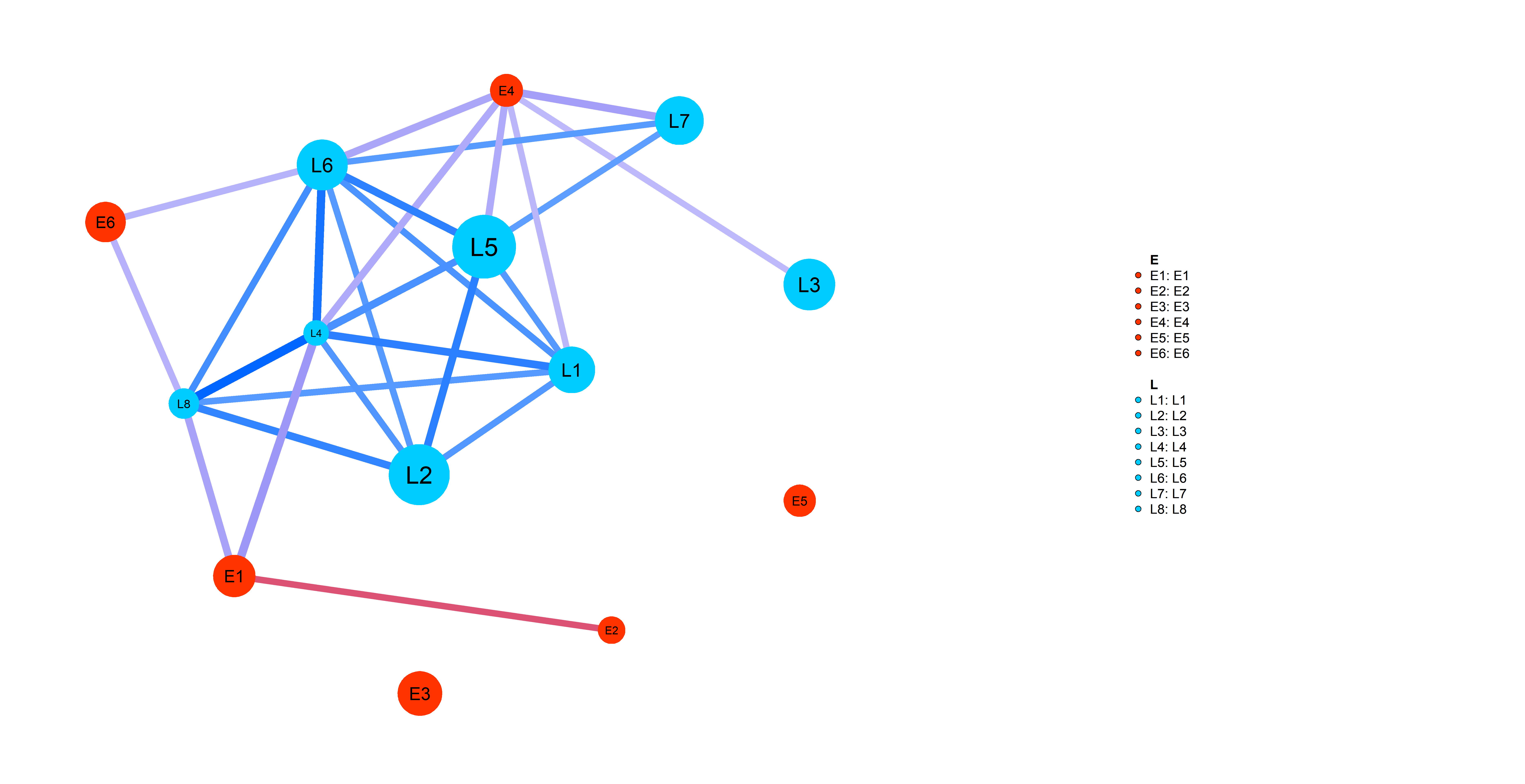
Simp_res=wrapper_human_mutations(geno,info,net,pathways_l,dataset_name,disease_type,key_words, n_cores=5,test_run=T,seed=0)Following plot shows you an example of pathway-specific patient similarity network between LIHC Late (L) stage cancer patients and Early (E)
MIT @ Giudice Luca