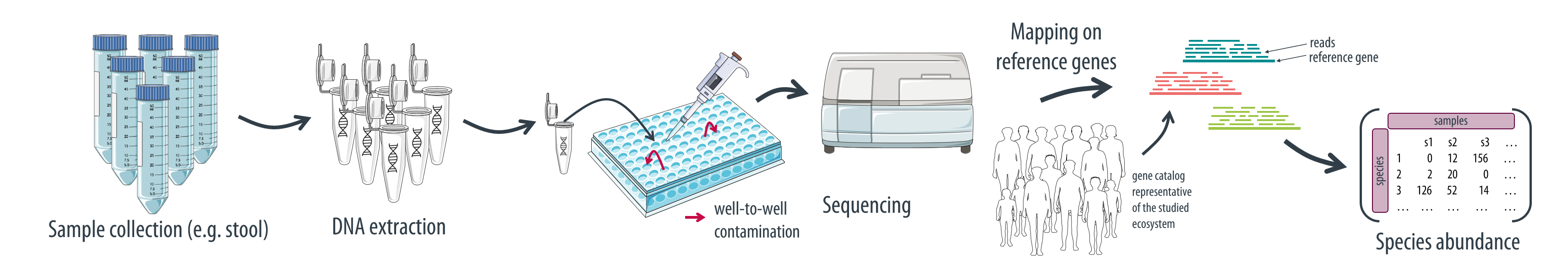
Cross‐sample contamination
Samples subjected to metagenomic sequencing can be accidentally contaminated during wet lab steps (DNA extraction, library preparation). Contamination refers to the presence of DNA that does not originate from the biological sample under study. It can be due either to:
- DNA from an external source (such as environmental DNA or lab reagents)
- DNA from another sample processed on the same plate (cross-sample/well-to-well contamination).
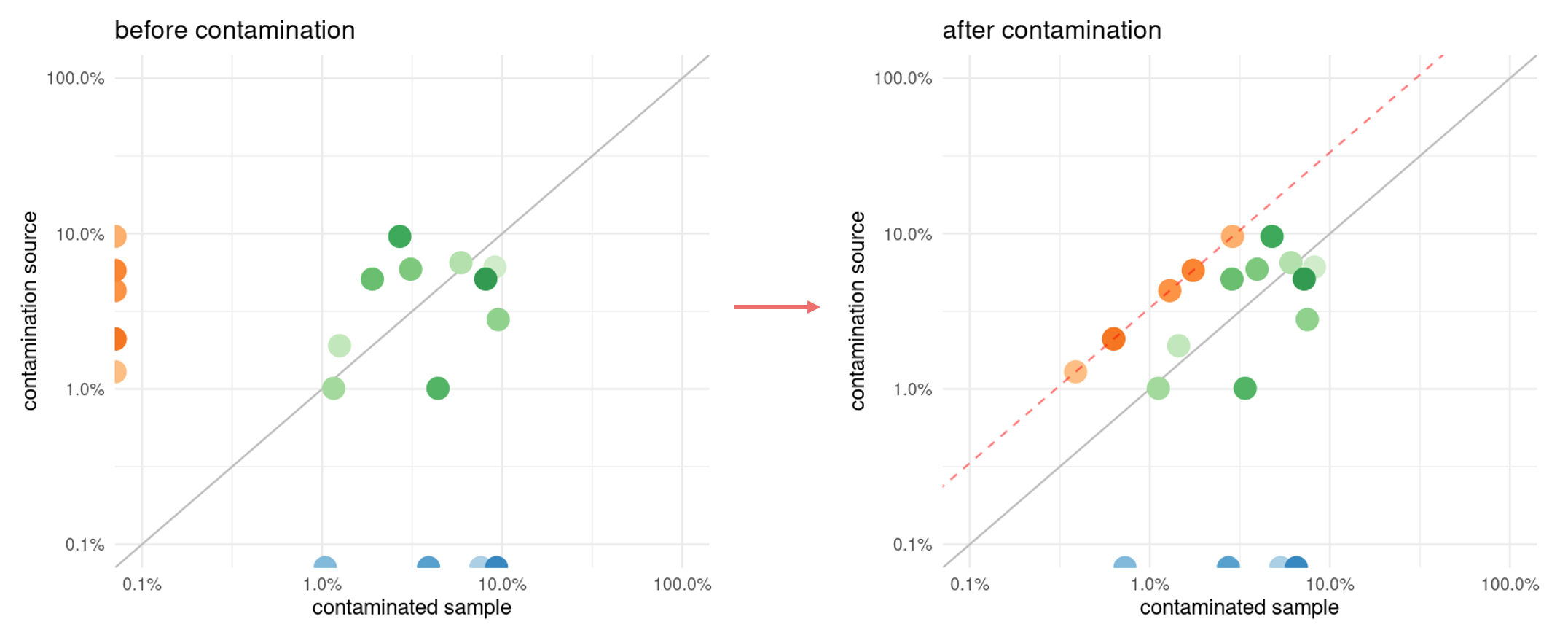
A species abundance profile is a representation of the abundance of species in one sample in relation to those in another. The profiles are on a log-log scale. Each point represents one species. Conventionally, the axes represent the species present in only one of the samples. By inspecting species abundance profiles of published cohort samples, we identified specific patterns associated with cross-sample contamination.
After contamination and above a certain threshold, all the abundant species of the contamination source sample are present in the contaminated sample. For a given species, its abundance in the contaminated sample is:
If the species was initially absent in the target sample (
Thus, these species have a proportional abundance between the two samples and form a contamination line.
The contamination line is used to detect contamination events, and the contamination rate can be estimated from relative abundance ratio of species that constitute it.