{kind=link}
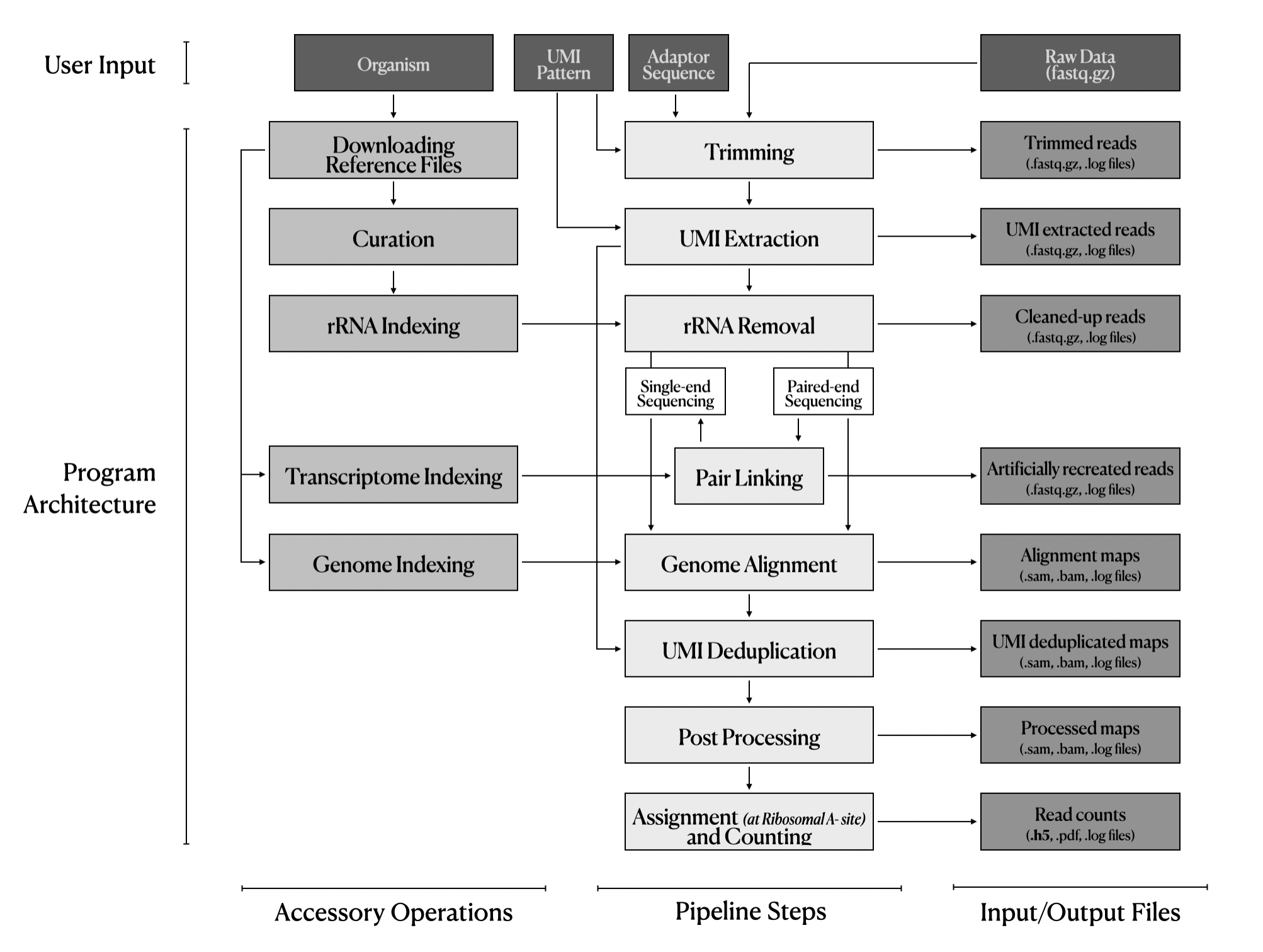
User provides these four inputs to the package. Then, the package starts running all necessary operations and modules. The package is composed of three main layers. accessory operations, actual pipeline steps, and input-output files. This one-click program accepts many raw FASTQ files at the same time. Auto-downloading all databases makes the analysis easy, reproducible, and up-to-date. Hyperparameters of each pipeline steps are optimized by randomized grid-search to get best quality results. This pipeline automates and standardizes analysis procedures. Some lab members have already started using this pipeline.
- Auto-downloading all necessary databases.
- Easy and reproducible analysis.
- Optimized hyperparameters.
- Automates and standardizes analysis procedures.
- Robust, rapid analysis for everyone.
>python3 pipeline.py -h
usage: pipeline.py [-h] [-r IDENTIFIER]
[-a {homo_sapiens,mus_musculus,saccharomyces_cerevisiae,escherichia_coli}]
[-e ENSEMBL_RELEASE] [-c CPU] [-s {3,5}] -f FILEPATH -t
TEMP -o OUTPUT
Deep sequencing data processing pipeline.
optional arguments:
-h, --help show this help message and exit
-r IDENTIFIER the identifier for this run, which will be the
directory name for all outputs under provided main
output directory. If skipped, the default is a string
containing date and time of the program start.
-a {homo_sapiens,mus_musculus,saccharomyces_cerevisiae,escherichia_coli}
organism of interest for the analysis. If skipped, the
default value is homo_sapiens.
-e ENSEMBL_RELEASE ensembl version to be used. Ignored if the 'organism'
does not necessitates Ensembl release information.
Default value is 102. For E. coli, it must be 48.
-c CPU number of cpu cores to be used. Default value is
maximum minus eight.
-s {3,5} select 3' or 5' assignment for Julia script.
-f FILEPATH path of the txt file which contains the task list.
-t TEMP absolute path of the directory to be used for
temporary files such as genome indexes.
-o OUTPUT absolute path of the directory to be used for output
files.
Example terminal command:
python3 /home/kai/HDD_Kemal/from_raf_computer/Kemal/RiboSeqProcess/pipeline.py -r JaroData -a escherichia_coli -e 48 -f /home/kai/HDD_Kemal/from_raf_computer/Kemal/RiboSeqProcess/example/jaro.txt -t /home/kai/HDD_Kemal/SequencingProcess/Temp/ -o /home/kai/HDD_Kemal/SequencingProcess/Output/
Example task list file can be found under example folder of this repository.
- It must be a
.txtdocument. - If a line starts with
#character, it will be interpreted as comments and disregarded. Empty lines are also ignored. - Sequencing files must be with
.fastq.gzextension. - All file paths have to be absolute
- A task should start with
>>>followed by a string (without spaces) which indicates the name of the sample. - Second line should be one of the followings: -"single" for single end sequencing -"paired" for paired end sequencing -"paired_linking" for paired end sequencing but also activates link pairing module.
- Composition of next lines depends on sequencing type.
-"single"
- File path for fastq file.
- Specific settings:
- "adapter": Adapter sequence
- "pattern_umi": Regex code for UMI extraction. -"paired" or "paired_linking"
- File path for fastq file for read 1 (forward).
- File path for fastq file for read 2 (reverse).
- Specific settings:
- "adapter1": Adapter sequence for read 1
- "adapter2": Adapter sequence for read 2
- "pattern_umi1": Regex code for UMI extraction for read 1
- "pattern_umi2": Regex code for UMI extraction for read 2
- About Specific settings: Order does not matter. If these are omitted, then the default values will be accepted. Key and value should be separated by
=character. For example,adapter=ATATATACG. Note thatNoneshould be written in order to omit such a settings; for example, if you do not want to cut anything, thenadapter=None. - Please see example task lists under
examplefolder.