

modlAMP
This is a Python package that is designed for working with peptides, proteins or any amino acid sequence of natural
amino acids. It incorporates several modules, like descriptor calculation (module descriptors) or sequence
generation (module sequences). For basic instructions how to use the package, see Usage section of this README
or the documentation.
Quick note: modlAMP supports Python 3 since version 4. Use with Python 2.7 is deprecated.
For the installation to work properly, pip needs to be installed. If you're not sure whether you already have pip,
type pip --version in your terminal. If you don't have pip installed, install it via sudo easy_install pip.
There is no need to download the package manually to install modlAMP. In your terminal, just type the following command:
pip install modlamp
To update modlamp to the latest version, run the following:
pip install --upgrade modlamp
This section gives a quick overview of different capabilities of modlAMP. For a detailed description of all modules see the module documentation.
After installation, you should be able to import and use the different modules like shown below. Type python or ipython in your terminal to begin, then the following import statements:
>>> from modlamp.sequences import Helices
>>> from modlamp.descriptors import PeptideDescriptor
>>> from modlamp.database import query_databaseThe following example shows how to generate a library of 1000 sequences out of all available sequence generation methods:
>>> from modlamp.sequences import MixedLibrary
>>> lib = MixedLibrary(1000)
>>> lib.generate_sequences()
>>> lib.sequences[:10]
['VIVRVLKIAA','VGAKALRGIGPVVK','QTGKAKIKLVKLRAGPYANGKLF','RLIKGALKLVRIVGPGLRVIVRGAR','DGQTNRFCGI','ILRVGKLAAKV',...]These commands generated a mixed peptide library comprising of 1000 sequences.
Note
If duplicates are present in the attribute sequences, these are removed during generation. Therefore it
is possible that less than the specified sequences are obtained.
The module sequences incorporates different sequence generation classes (random, helices etc.). For
documentation thereof, consider the docs for the module modlamp.sequences.
Now, different descriptor values can be calculated for the generated sequences: (see Generating Sequences)
How to calculate the Eisenberg hydrophobic moment for given sequences:
>>> from modlamp.descriptors import PeptideDescriptor, GlobalDescriptor
>>> desc = PeptideDescriptor(lib.sequences,'eisenberg')
>>> desc.calculate_moment()
>>> desc.descriptor[:10]
array([[ 0.60138255],[ 0.61232763],[ 0.01474009],[ 0.72333858],[ 0.20390763],[ 0.68818279],...]Global descriptor features like charge, hydrophobicity or isoelectric point can be calculated as well:
>>> glob = GlobalDescriptor(lib.sequences)
>>> glob.isoelectric_point()
>>> glob.descriptor[:10]
array([ 10.09735107, 8.75006104, 12.30743408, 11.26385498, ...]Auto- and cross-correlation type functions with different window sizes can be applied on all available amino acid scales. Here an example for the pepCATS scale:
>>> pepCATS = PeptideDescriptor('sequence/file/to/be/loaded.fasta', 'pepcats')
>>> pepCATS.calculate_crosscorr(7)
>>> pepCATS.descriptor
array([[ 0.6875 , 0.46666667, 0.42857143, 0.61538462, 0.58333333,Many more amino acid scales are available for descriptor calculation. The complete list can be found in the
documentation for the modlamp.descriptors module.
We can also plot the calculated values as a boxplot, for example the hydrophobic moment:
>>> from modlamp.plot import plot_feature
>>> D = PeptideDescriptor('sequence/file/to/be/loaded.fasta', 'eisenberg') # Eisenberg hyrophobicity scale
>>> D.calculate_moment()
>>> plot_feature(D.descriptor,y_label='uH Eisenberg')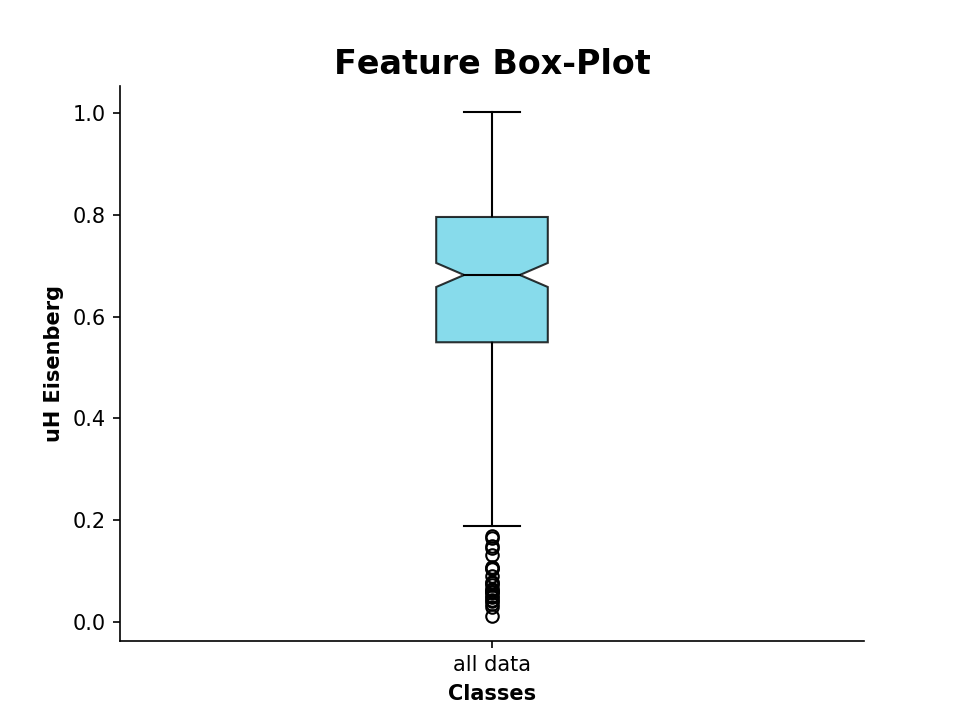
We can additionally compare these descriptor values to known AMP sequences. For that, we import sequences from the
APD3, which are stored in the FASTA formatted file APD3.fasta:
>>> APD = PeptideDescriptor('/Path/to/file/APD3.fasta', 'eisenberg')
>>> APD.calculate_moment()Now lets compare the values by plotting:
>>> plot_feature([D.descriptor, APD.descriptor], y_label='uH Eisenberg', x_tick_labels=['Library', 'APD3'])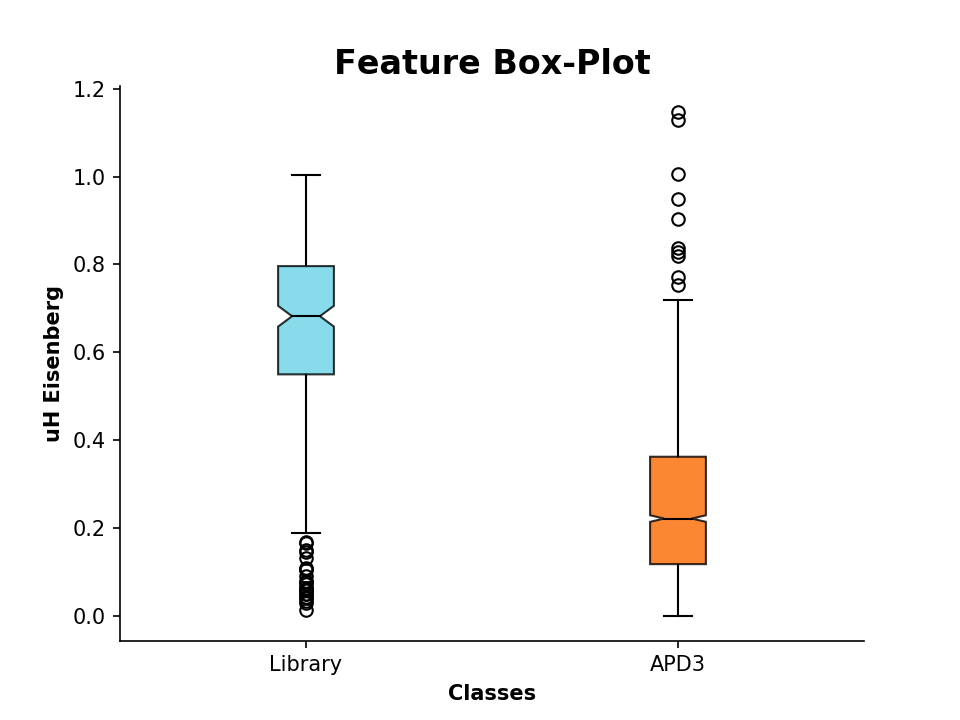
It is also possible to plot 2 or 3 different features in a scatter plot:
| Example: | 2D Scatter Plot |
|---|
>>> from modlamp.plot import plot_2_features
>>> A = PeptideDescriptor('/Path/to/file/class1&2.fasta', 'eisenberg')
>>> A.calculate_moment()
>>> B = GlobalDescriptor('/Path/to/file/class1&2.fasta')
>>> B.isoelectric_point()
>>> target = [1] * (len(A.sequences) / 2) + [0] * (len(A.sequences) / 2)
>>> plot_2_features(A.descriptor, B.descriptor, x_label='uH', y_label='pI', targets=target)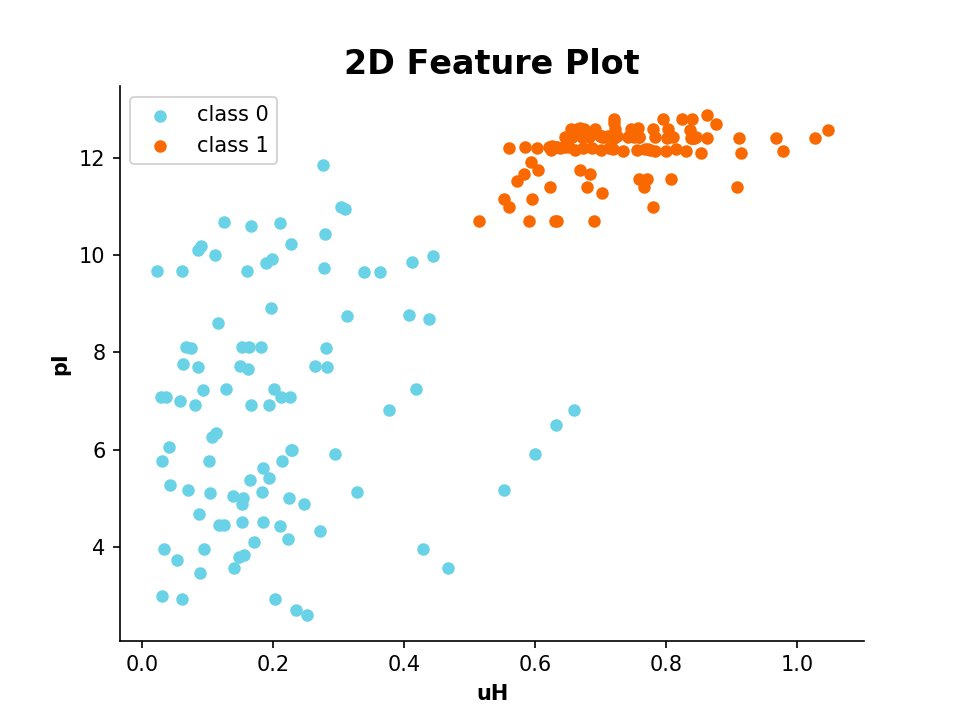
| Example: | 3D Scatter Plot |
|---|
>>> from modlamp.plot import plot_3_features
>>> B = GlobalDescriptor(APD.sequences)
>>> B.isoelectric_point()
>>> B.length(append=True) # append descriptor values to afore calculated
>>> plot_3_features(APD.descriptor, B.descriptor[:, 0], B.descriptor[:, 1], x_label='uH', y_label='pI', z_label='len')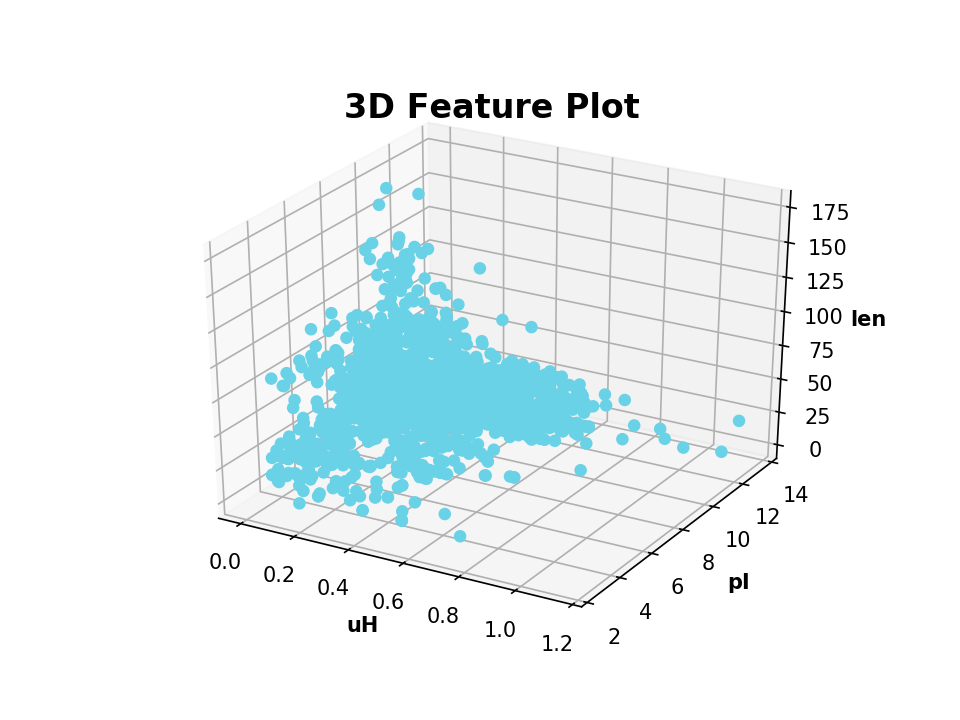
| Example: | Helical Wheel Plot |
|---|
>>> from modlamp.plot import helical_wheel
>>> helical_wheel('GLFDIVKKVVGALGSL', moment=True)
Further plotting methods are available. See the documentation for the modlamp.plot
module.
Peptides from the two most prominent AMP databases APD and CAMP can be directly scraped with the modlamp.database module.
For downloading a set of sequences from the APD database, first get the IDs of the sequences you want to query from the APD website. Then proceed as follows:
>>> query_apd([15, 16, 17, 18, 19, 20]) # download sequences with APD IDs 15 to 20
['GLFDIVKKVVGALGSL','GLFDIVKKVVGAIGSL','GLFDIVKKVVGTLAGL','GLFDIVKKVVGAFGSL','GLFDIAKKVIGVIGSL','GLFDIVKKIAGHIAGSI']The same holds true for the CAMP database:
>>> query_camp([2705, 2706]) # download sequences with CAMP IDs 2705 & 2706
['GLFDIVKKVVGALGSL','GLFDIVKKVVGTLAGL']modlAMP also hosts a module for connecting to your own database on a private server. Peptide sequences included in any table in the database can be downloaded.
Note
The modlamp.database.query_database function allows connection and queries to a personal database. For
successful connection, the database configuration needs to be specified in the db_config.json file, which is
located in modlamp/data/ by default.
Sequences (stored in a column named sequence) from the personal database can then be queried as follows:
>>> from modlamp.database import query_database
>>> query_database('my_experiments', ['sequence'], configfile='./modlamp/data/db_config.json')
Password: >? ***********
Connecting to MySQL database...
connection established!
['ILDSSWQRTFLLS','IKLLHIF','ACFDDGLFRIIKFLLASDRFFT', ...]For AMP QSAR models, different options exist of choosing negative / inactive peptide examples. We assembled several
datasets for classification tasks, that can be read by the modlamp.datasets module. The available datasets can
be found in the documentation of the modlamp.datasets module.
| Example: | AMPs vs. transmembrane regions of proteins |
|---|
>>> from modlamp.datasets import load_AMPvsTM
>>> data = load_AMPvsTM()
>>> data.keys()
['target_names', 'target', 'feature_names', 'sequences']The variable data holds four different keys, which can also be called as its attributes. The available
attributes for load_helicalAMPset() are target_names (target names), target (the
target class vector), feature_names (the name of the data features, here: 'Sequence') and
sequences (the loaded sequences).
| Example: |
|---|
>>> data.target_names # class names
array(['TM', 'AMP'], dtype='|S3')
>>> data.sequences[:5] # sequences
[array(['AAGAATVLLVIVLLAGSYLAVLA', 'LWIVIACLACVGSAAALTLRA', 'FYRFYMLREGTAVPAVWFSIELIFGLFA', 'GTLELGVDYGRAN',
'KLFWRAVVAEFLATTLFVFISIGSALGFK'], dtype='|S100')
>>> data.target # corresponding target classes
array([0, 0, 0, 0, 0 .... 1, 1, 1, 1])The modlule modlamp.wetlab includes the class modlamp.wetlab.CD to analyse raw circular dichroism
data from wetlab experiments. The following example shows how to load a raw datafile and calculate secondary
structure contents:
>>> cd = CD('/path/to/your/folder', 185, 260) # load all files in a specified folder
>>> cd.names # peptide names read from the file headers
['Pep 10', 'Pep 10', 'Pep 11', 'Pep 11', ... ]
>>> cd.calc_meanres_ellipticity() # calculate the mean residue ellipticity values
>>> cd.meanres_ellipticity
array([[ 260. , -266.95804196],
[ 259. , -338.13286713],
[ 258. , -387.25174825], ...])
>>> cd.helicity(temperature=24., k=3.492185008, induction=True) # calculate helical content
>>> cd.helicity_values
Name Solvent Helicity Induction
Peptide1 T 100.0 3.823
Peptide1 W 26.16 0.000
Peptide2 T 76.38 3.048
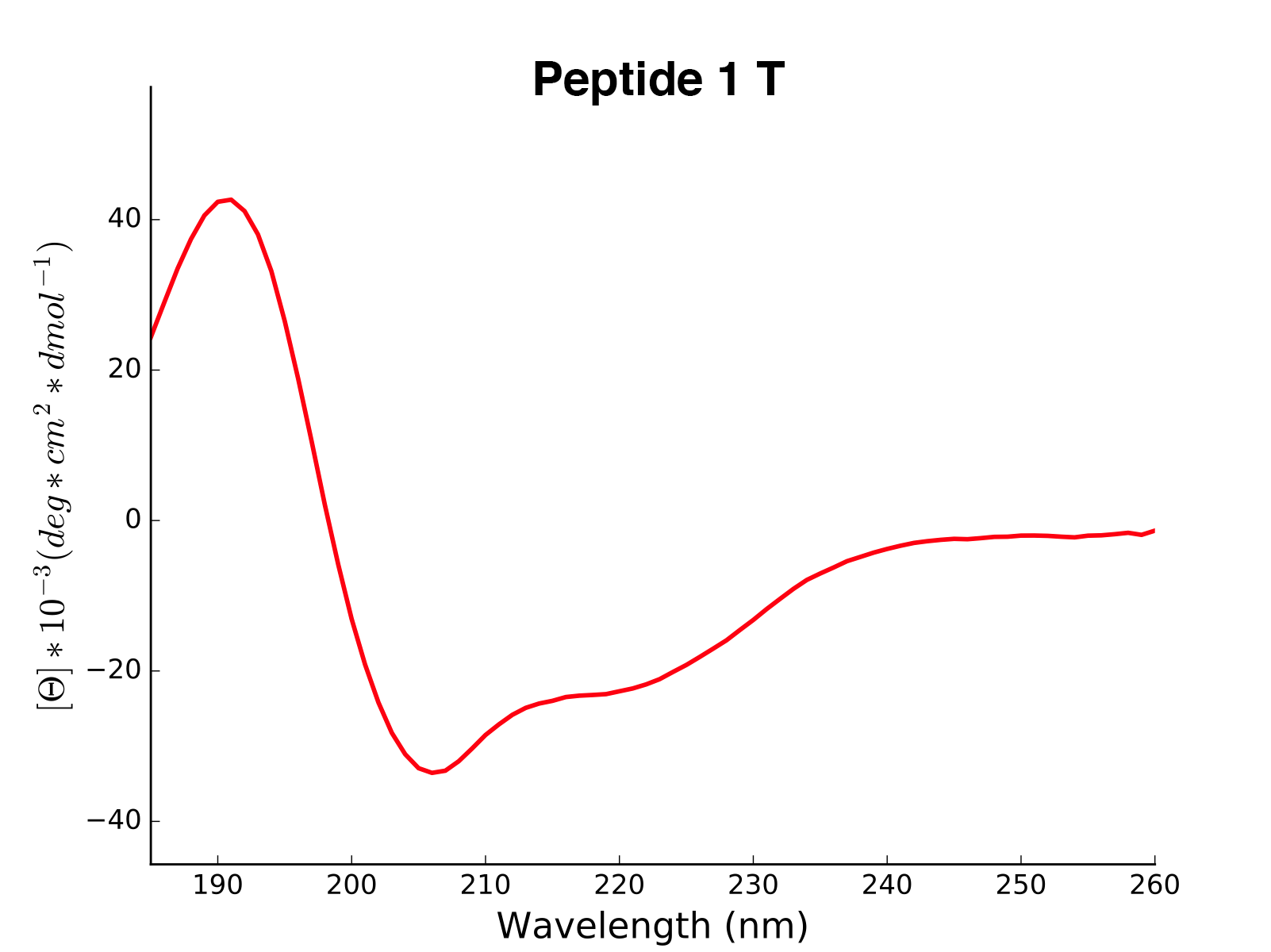
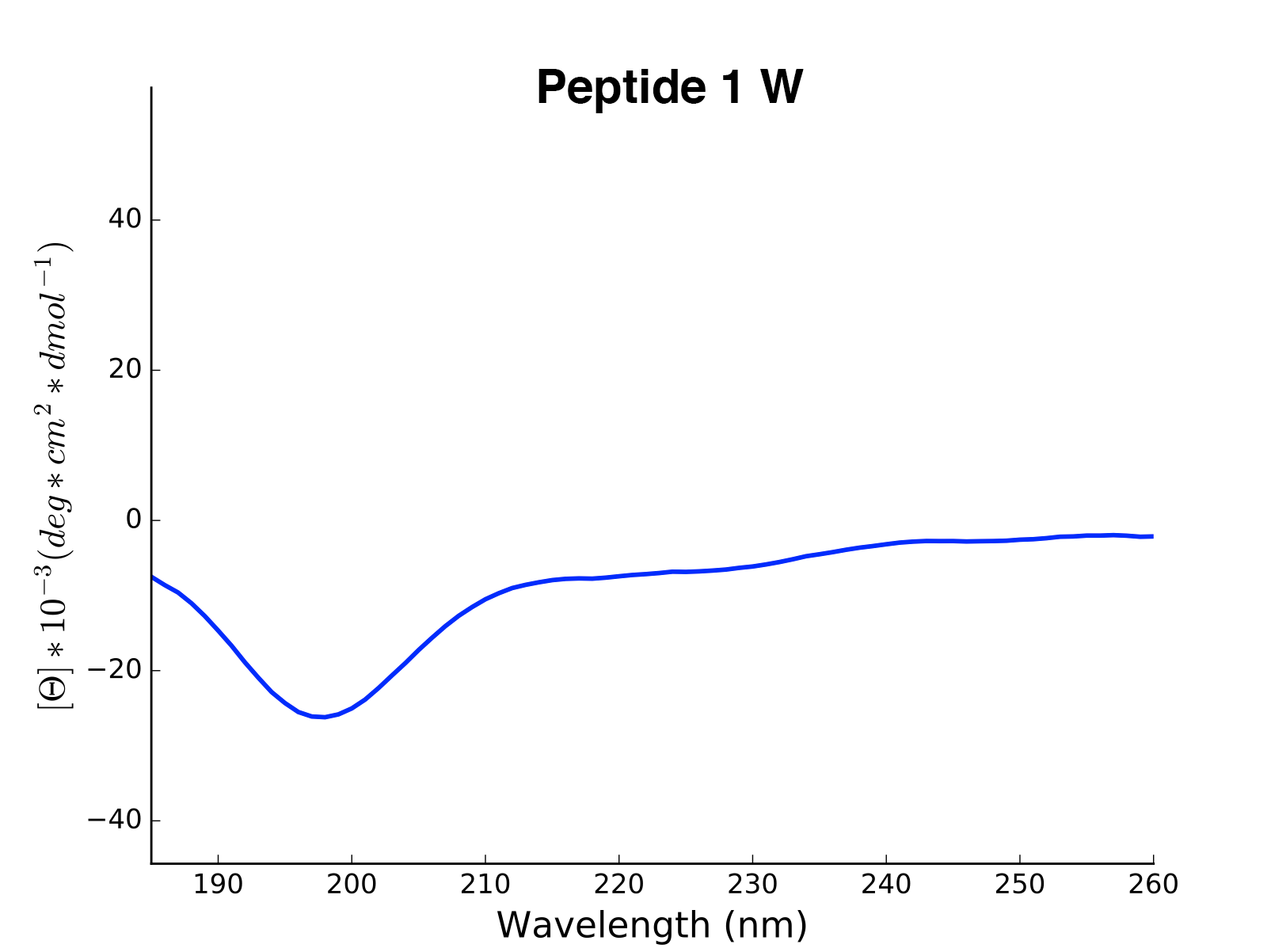
Peptide2 W 25.06 0.000 ...The read and calculated values can finally be plotted as follows:
>>> cd.plot(data='mean residue ellipticity', combine=True)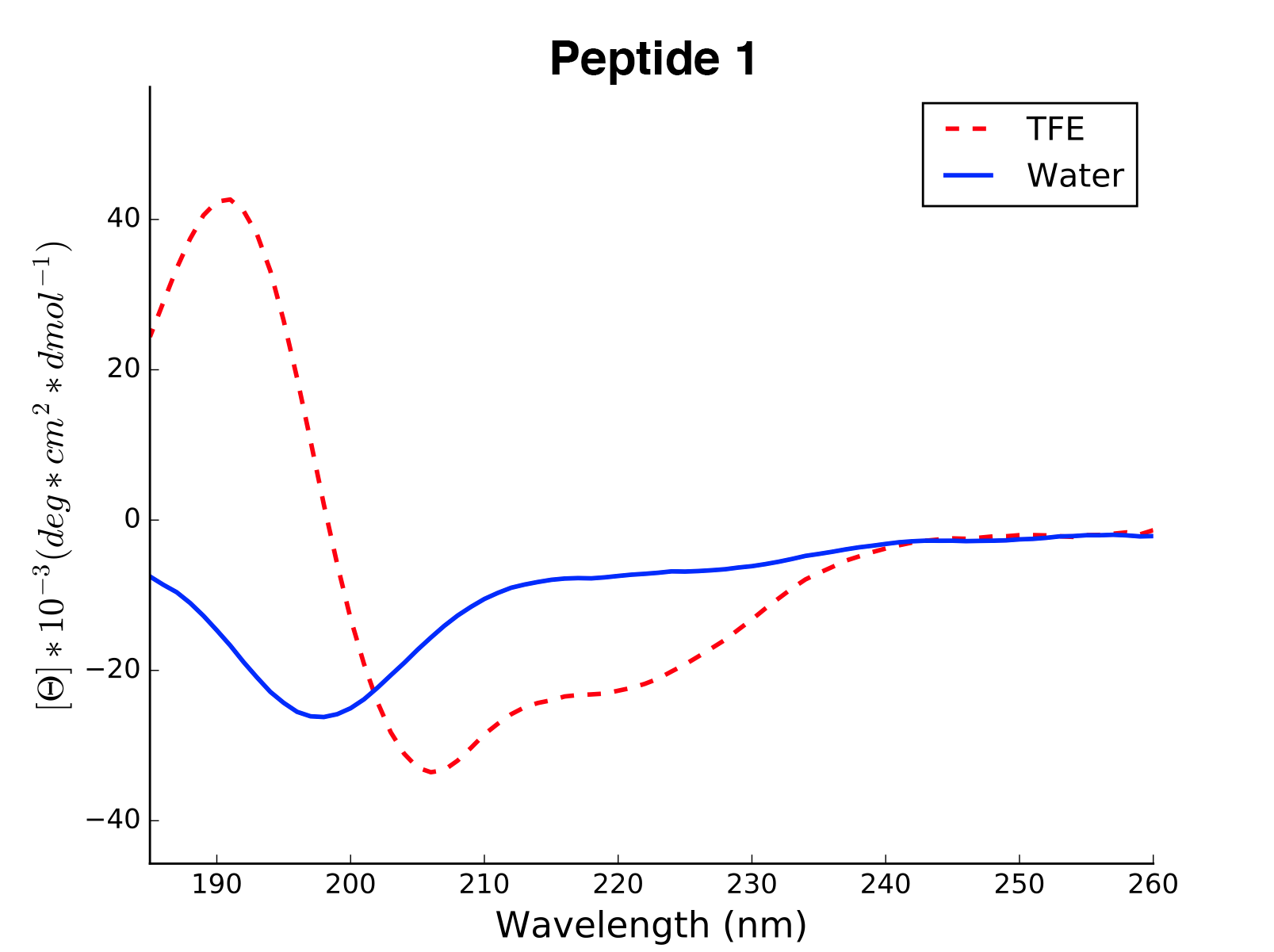
The modlule modlamp.analysis includes the class modlamp.analysis.GlobalAnalysis to compare
different sequence libraries. Learn how to use it with the following example:
>>> lib # sequence library with 3 sub-libraries
array([['ARVFVRAVRIYIRVLKAFAKL', 'IRVYVRIVRGFGRVVRAYARV', 'IRIFIRIARGFGRAIRVFVRI', ..., 'RGPCFLQVVD'],
['EYKIGGKA', 'RAVKGGGRLLAG', 'KLLRIILRGARIIIRGLR', ..., 'AKCLVDKK', 'VGGAFALVSV'],
['GVHLKFKPAVSRKGVKGIT', 'RILRIGARVGKVLIK', 'MKGIIGHTWKLKPTIPSGKSAKC', ..., 'GRIIRLAIKAGL']], dtype='|S28')
>>> lib.shape
(3, 2000)
>>> from modlamp.analysis import GlobalAnalysis
>>> analysis = GlobalAnalysis(lib, names=['Lib 1', 'Lib 2', 'Lib 3'])
>>> analysis.plot_summary()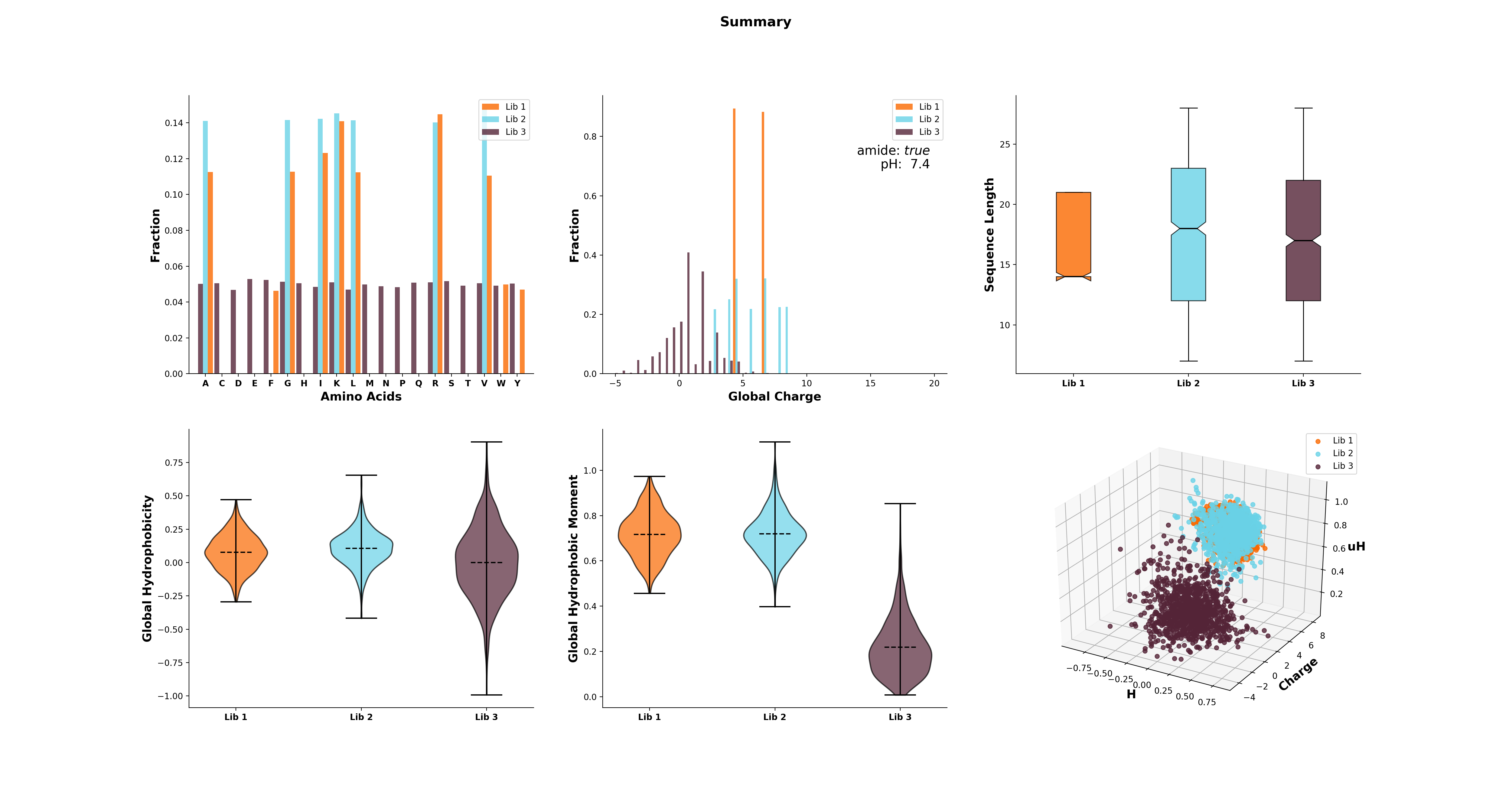
A detailed documentation of all modules is available from the modlAMP documentation website.
If you are using modlAMP for a scientific publication, please cite the following paper:
Müller A. T. et al. (2017) modlAMP: Python for anitmicrobial peptides, Bioinformatics 33, (17), 2753-2755, DOI:10.1093/bioinformatics/btx285.