Introduction
Multiplex sequencing allows for many samples to be sequenced in a single run, saving time and money. Unique index sequences are added to each sample library so that the sample identity of each read can be determined. Many sets of indexes are commercially available, but these become quite costly for large numbers of samples.
To lower the cost of multiplex library preparation, we designed a program that generates primers containing compatible indexes, which can be ordered as standard oligos for a fraction of the cost of commercially-available indexing primers. The program outputs primer sequences in a ready-to-order format and also generates sample sheets to use for demultiplexing.
By default, GIL generates primers compatible with Illumina TruSeq. This explanation focuses on TruSeq, but note that you can generate primers for Nextera libraries or even custom library types if desired.
An Illumina TruSeq dual-indexed library has the following structure (adapted from the Teichmann Lab's GitHub, a great resource for understanding how Illumina sequencing works):
5'- AATGATACGGCGACCACCGAGATCTACACNNNNNNNNACACTCTTTCCCTACACGACGCTCTTCCGATCT-insert-AGATCGGAAGAGCACACGTCTGAACTCCAGTCACNNNNNNNNATCTCGTATGCCGTCTTCTGCTTG -3'
3'- TTACTATGCCGCTGGTGGCTCTAGATGTGNNNNNNNNTGTGAGAAAGGGATGTGCTGCGAGAAGGCTAGA-insert-TCTAGCCTTCTCGTGTGCAGACTTGAGGTCAGTGNNNNNNNNTAGAGCATACGGCAGAAGACGAAC -5'
Illumina P5 i5 Truseq Read 1 Truseq Read 2 i7 Illumina P7
The P5 and P7 sequences bind to the flow cell, and the Read 1 and Read 2 sequences bind to sequencing primers.
To add indexes to a library, inserts must be flanked with Read 1/Read 2 sequences. These sequences can be added using ligation: e.g. NEBNext Adaptor for Illumina, or the "stubby Y-yoke adaptor" from the iTru Library Method. These sequences can also be added by PCR for sequencing inserts from a specific locus, such as in MPRA experiments. The following diagram shows how our indexing primers anneal to a library of inserts flanked with TruSeq Read 1/Read 2 sequences. The complementary part of the primers was designed for a Tm of 65 °C with Q5 polymerase.
5'- AATGATACGGCGACCACCGAGATCTACACNNNNNNNNACACTCTTTCCCTACACGACG -3'
5'- ACACTCTTTCCCTACACGACGCTCTTCCGATCT-insert-AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC -3'
3'- TGTGAGAAAGGGATGTGCTGCGAGAAGGCTAGA-insert-TCTAGCCTTCTCGTGTGCAGACTTGAGGTCAGTG -5'
3'- GTGCAGACTTGAGGTCAGTGNNNNNNNNTAGAGCATACGGCAGAAGACGAAC -5'
Illumina P5 i5 Truseq Read 1 Truseq Read 2 i7 Illumina P7
GIL starts by generating a list of all possible indexes of the specified length (by default, all possible 8mers). For indexes of length > 9, a random sample of indexes is used, as using all possible kmers would take too long.
In sequencers that use 2-channel chemistry (e.g. NovaSeq 6000), G is not labelled with a fluorescent dye and the presence of G is inferred from a lack of signal. Illumina warns that if the first two bases in the index are both G, "signal intensity is not generated."
To ensure that signal intensity is generated for every index, we remove any indexes that start with G. For the i5 (read 2) index we also remove any indexes with a reverse complement that begins with G, as this index may be read in either direction depending on the sequencer used.
For a less stringent filtering, only sequences beginning with GG (and i5 ending with CC) are removed. This filtering typically results in five plates of indexes instead of three (with all default parameters otherwise).
Indexes with extreme GC content could lead to differences in PCR efficiency between different primers. GIL removes indexes with GC content less than 25% or greater than 75% by default.
Homopolymer and dinucleotide repeats can cause issues such as polymerase stuttering and mispriming. Runs of G can also cause issues during PCR due to intermolecular stacking. We decided to remove indexes with >2 homopolymer or dinucleotide repeats by default to avoid these potential issues.
If an index is complementary to the 3' end of the primer, the 3' end of the primer could fold back onto itself and be extended by the polymerase, ruining the primer. To prevent this issue, we compute the Hamming distance between the reverse complement of the 8 bases on the 3' end of the primer and all 8 nt windows that include the index sequence. If any of these distances are >3, the index is filtered out.
After filtering, we choose a subset of the remaining sequences
such that no two sequences are too similar. To calculate similarity we use
Levenshtein distance, which is the number
of substitutions, insertions, or deletions required to change one sequence to the other sequence.
We chose Levenshtein distance because it accounts for deletions, which are the most
common error in synthetic oligos. The Levenshtein module
distance function, which is implemented in C, is used by default to calculate distance. If this
module isn't available, a much slower Python implementation of Levenshtein distance is used.
Before selecting the set of dissimilar sequences, the indexes can optionally be filtered to remove sequences that are
too similar to an existing set of indexes. For example, if you will be using these indexes
alongside commercial indexes such as NEBNext® Multiplex Oligos for Illumina®, you can ensure
that none of the generated indexes are too similar by using the --blocklist option along with
a file containing the indexes. A more detailed explanation of this option can be found in the
usage examples section.
To select this dissimilar set, we first select a random sequence from the set of filtered indexes, then remove all sequences that are too similar to this sequence. We then repeat this cycle of selecting a new sequence and removing all similar sequences until there are no more indexes left to select from. This does not necessarily generate the largest possible set of sequences, but it is much faster than trying to find this optimal set of dissimilar sequences and with default settings we routinely get three 96-well plates of indexes.
When only a few indexes are used in a sequencing run, colour balance must be considered. Most Illumina sequencers use one of two main technologies for imaging: 2-channel and 4-channel. A detailed explanation of why colour balance is important can be found here. In short, during each sequencing cycle, multiple images are taken using different wavelengths of light, as the fluorescent dyes attached to the bases are excited at different wavelengths. If all of the bases at a certain position in the indexes are imaged in the same channel, the other channel will be completely dark, which can cause the run to fail. It is therefore important that the correct diversity of bases is present at each position in the set of indexes used in a run.
The program outputs indexes such that each group of four indexes along the rows of the plate is colour balanced. This allows for multiplexing of as few as four indexes without having to worry about colour balance.
Two indexing strategies are possible with dual indexes. Unique dual indexing (UDI) uses a unique read 1 index and a unique read 2 index for each sample. As the number of samples increases, this can get expensive, as two new indexing primers are needed for each new sample. Combinatorial dual indexing (CDI) solves this problem by using indexes multiple times while ensuring that each pair of indexes is unique. Using CDI, only 20 primers are needed to index 96 samples, while 192 primers would be required with UDI.
An illustration of CDI vs UDI from the Illumina website:
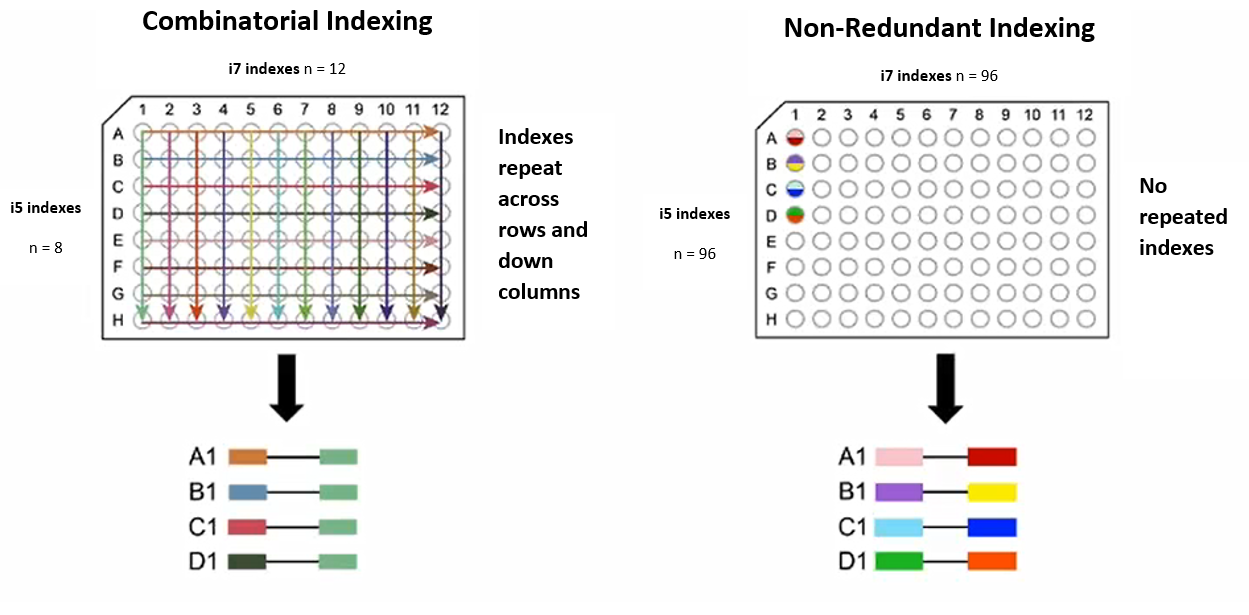
Why use UDI then? Something called index hopping can occur during sequencing, especially on certain sequencers. Index hopping is when one of the indexes from one sample occurs in a different sample. If this happens with UDIs, the index pairing will be invalid and the read can be thrown out. With CDIs, however, because indexes are shared between samples, index hopping could lead to an index combination that is still valid. This read would then be assigned to the wrong sample.
By default, GIL creates plates for the UDI indexing strategy: 96 i7 and 96 i5 primers for each plate generated. One set of 96 primer pairs for UDI can be expanded to 64 plates of CDI primers, which can index 6,144 samples. To do this, simply choose one row from the i7 plate and one row from the i5 plate. The 12 i7 indexes from the row are copied down the columns of the new plate, and the first 8 indexes of the i5 row are copied along the rows of the new plate.
GIL can create sample sheets for CDI plates that are constructed in this manner. For a detailed explanation, see this example.