Signals pertaining to transcriptional activation are transferred from enhancers found in intergenic loci to genes in the form of transcription factors, cofactors, and various transcriptional machineries such as RNA Pol II. How and where this information is transmitted to and from is central for decoding the regulatory landscape of any gene and identifying enhancers. Esearch3D is an unsupervised algorithm to predict enhancers. It reverse engineers the flow of information and identifies intergenic regulatory enhancers using solely gene expression and 3D genomic data. It models chromosome conformation capture (3C) data as chromatin interaction network (CIN) and then exploits graph-theory algorithms to integrate RNA-seq data to calculate an imputed activity score (IAS) for intergenic regions. We also provide a visualisation tool to allow an easy interpretation of the results.
![]()
- It uses the expression levels of genes and a chromatin interaction network to impute the activity score of enhancers represented as nodes
- It reverse engineers the flow of information modeled by enhancers that act as regulatory sources to increase the rate of transcription of their target genes
- It leverages the relationship between the 3D organisation of chromatin and global gene expression
- It represents a novel enhancer associated feature that can be used to predict enhancers
- It provides a graphical user interface to interact with the network composed by genes, enhancers and intergenic loci associated to their imputed activity score
- It includes a parallelized version of the network-based algorithm called random walk with restart that works on sparse matrices to reduce the system usage
Maninder Heer*, Luca Giudice*, Claudia Mengoni, Rosalba Giugno^, and Daniel Rico^. Esearch3D: Propagating gene expression in chromatin networks to illuminate active enhancers
*equally contributed ^co-senior authors
Esearch3D is available on R, you can install it by:
install.packages("devtools")
install.packages("htmltools", version="0.5.2", type="source")
devtools::install_github(
repo="InfOmics/Esearch3D",
ref = "main",
dependencies = "Depends",
upgrade = "always",
quiet = T
# type = "binary" # usable only in Windows OS
)There are the following vignettes:
- Vignette for IAS score computation on mouse embryonic stem cells (mESCs) DNaseI capture Hi-C dataset
- Vignette for IAS score computation and enhancer classification on mouse embryonic stem cells (mESCs) DNaseI capture Hi-C dataset
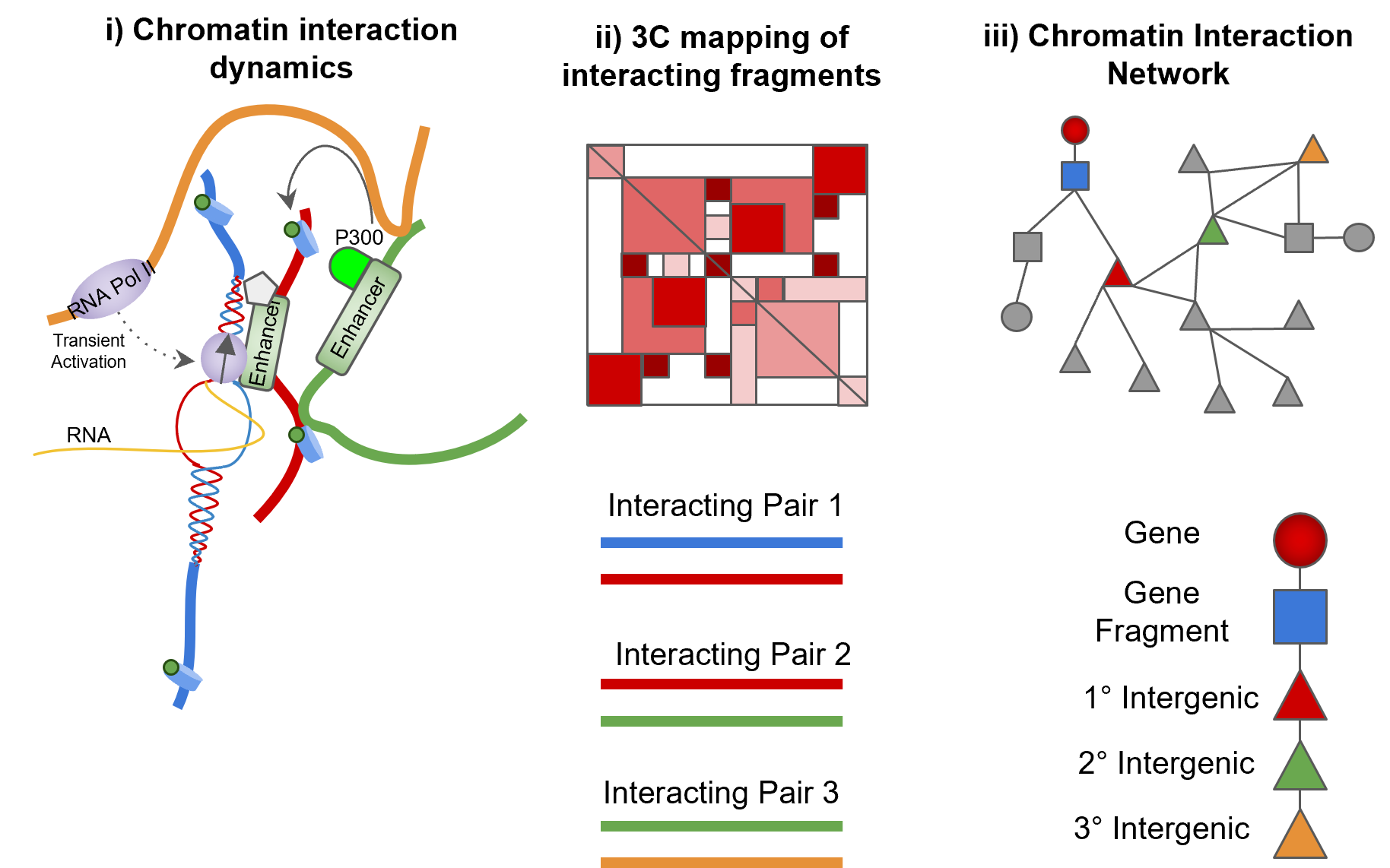
- Enhancers are localised and can influence the regulation of promoters directly and indirectly.
- 3C can capture chromatin interactions but only in a pairwise manner.
- Representation of chromatin fragments as nodes in a network and their interactions as edges preserves indirect associations between interacting chromatin.
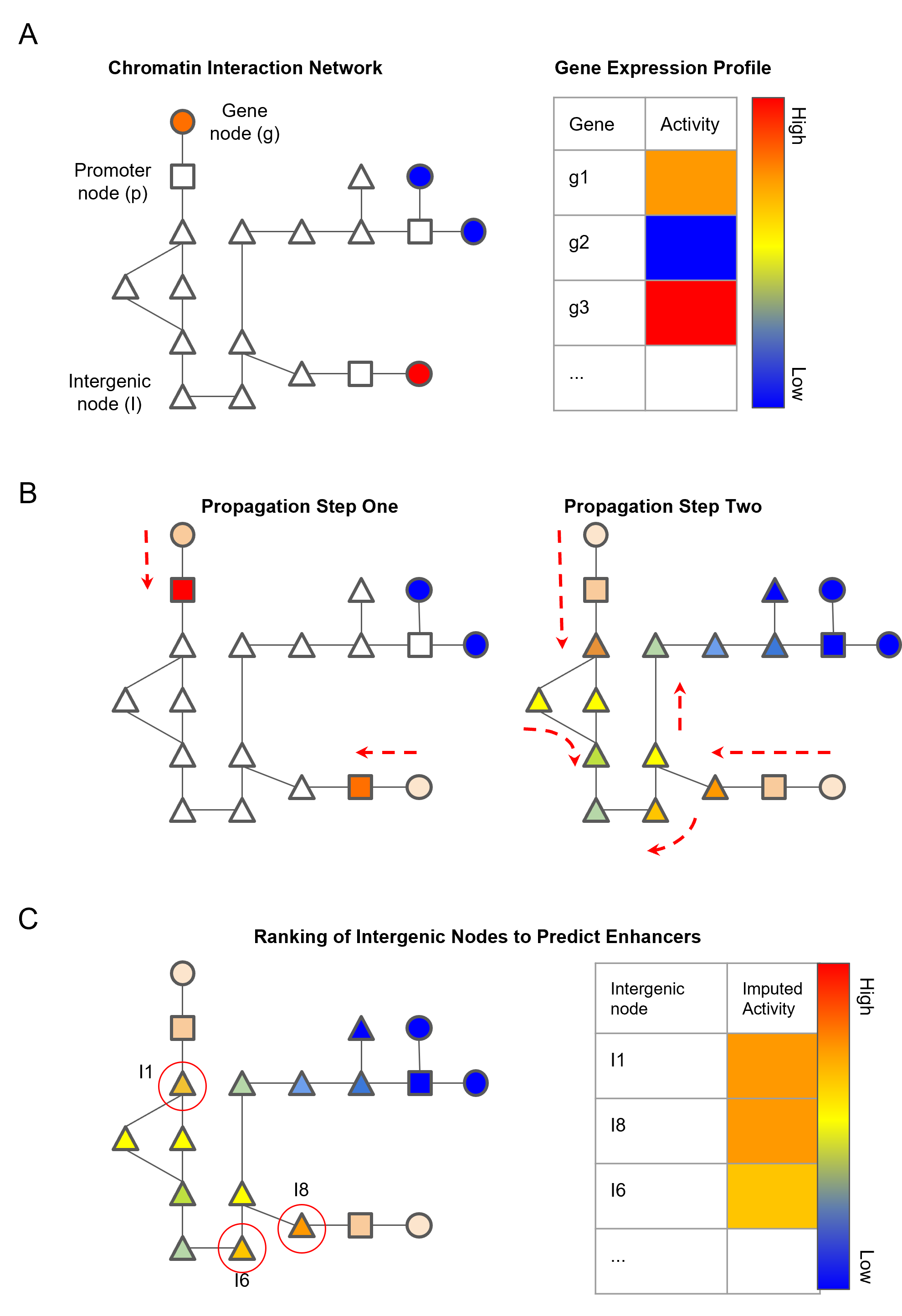
Schematic diagram of the network propagation used to impute activity values at intergenic nodes:
- A. Genes are mapped to nodes representing genic chromatin fragments. Each gene has an associated gene activity value determined by RNA-seq data.
- B. Gene activity is propagated from gene nodes to genic chromatin nodes in propagation step one. Activity scores are then imputed in intergenic chromatin nodes by propagating the scores from genic chromatin nodes.
- C. Ranking of non-genic nodes by the imputed activity score to identify high confidence enhancer nodes.
Few lines of code to run ESearch3D prediction:
library(Esearch3D)
#Load and set up the example data ----
data("wg_data_l")
#gene - fragment interaction network
gf_net=wg_data_ll$gf_net
#gene-fragment-fragment interaction network
ff_net=wg_data_l$ff_net
#sample profile with starting values for genes and fragments
input_m=wg_data_l$input_m
#length of chromosomes
chr_len=wg_data_l$chr_len
#gene annotation
ann_net_b=wg_data_l$ann_net_b
#Two step propagation -----
#Propagation over the gene-fragment network
gf_prop=rwr_OVprop(g=gf_net,input_m = input_m, no_cores=2, r=0.1)
#Propagation over the gene-fragment-fragment network
ff_prop=rwr_OVprop(g=ff_net,input_m = gf_prop, no_cores=2, r=0.8)
#Create igraph object with all the information included
net=create_net2plot(gf_net,input_m,gf_prop,ann_net_b,frag_pattern="frag",ff_net,ff_prop)
#Start GUI
start_GUI(net, ann_net_b)The GUI allows to explore a sample's profile after a network-based propagation. The user can investigate the imputed activity scores obtained by specific genes and their neighborhood. The visualized network can be downloaded and further analysed in Cytoscape.

- Type a node + Distance from a selected node: allows to visualize the neighbourhood of one specific node of interest
- Scale colours: allows to scale the colors of the visualized nodes as if the propagation would have been applied only on them
- Select by propagation ranges: it shows only the nodes with a value that falls inside a specific range
- Download html file: allows to download the network as an html file the network created with the GUI
- Download GML file: allows to download the network and its features as a GML file that can be opened in Cytoscape.
The following image shows you an example of how to interact with the GUI and its functionalities
