


{kind=link}
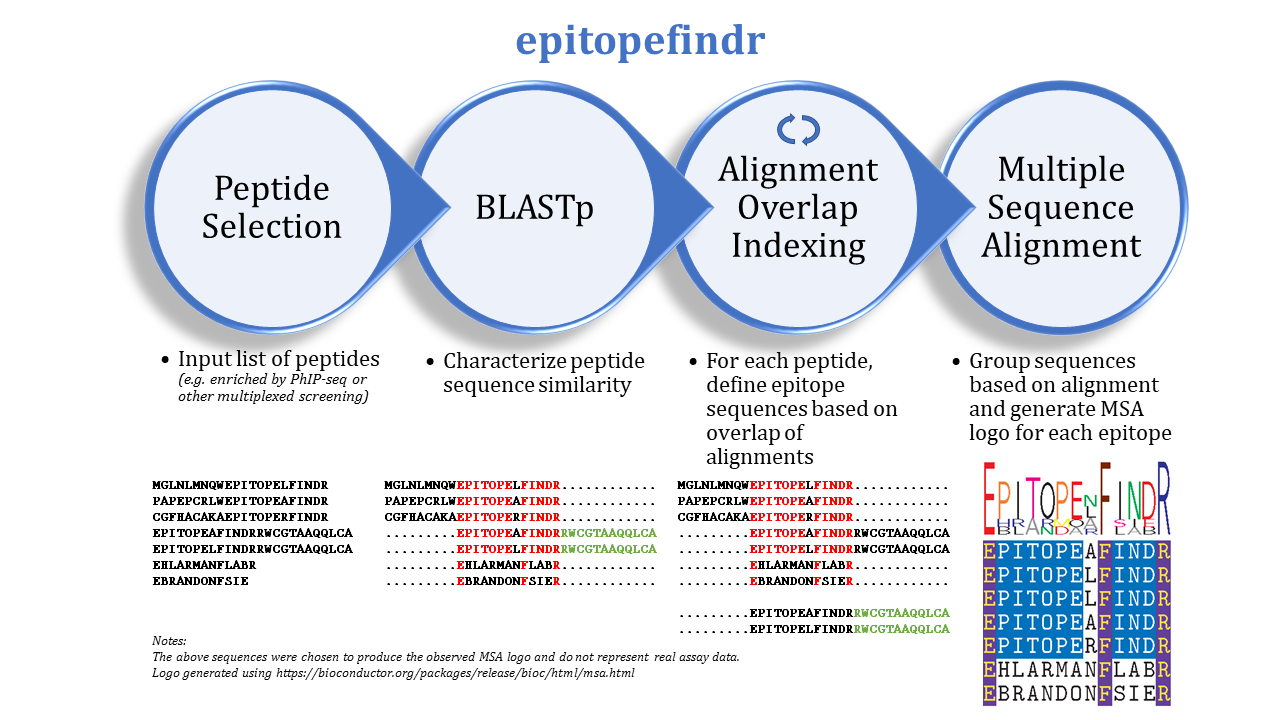
The purpose of this package is to describe the BLAST alignments among a set of peptide sequences by reporting the overlaps of each peptide's alignments to other peptides in the set. One can imagine inputting a list of peptides enriched by immunoprecipitation (e.g. by PhIP-seq) to identify corresponding epitopes.
epitopefindr takes a .fasta file listing peptide sequences of interest and calls BLASTp from within R to identify alignments among these peptides. Each peptide's alignments to other peptides are then simplified to the minimal number of "non overlapping" intervals* of the index peptide that represent all alignments to other peptides reported by BLAST. (*By default, each interval must be at least 7 amino acids long, and two intervals are considered NOT overlapping if they share 6 or fewer amino acids). After the minimal overlaps are identified for each peptide, these overlaps are gathered into aligning groups based on the initial BLAST. For each group, a multiple sequence alignment logo (motif) is generated to represent the collective sequence. Additionally, a spreadsheet is written to list the final trimmed amino acid sequences and some metadata.
- Install R (version 3.5+).
- Install BLAST+ (version 2.7.1). (Note: we have observed some issues with more recent versions of BLAST+ and will monitor for bugfixes.)
- In R console, execute:
if (!requireNamespace("devtools")) install.packages("devtools")
devtools::install_github("brandonsie/epitopefindr")
library(epitopefindr)(These are not essential to epitopefindr, but are used to generate alignment logo PDFs from the alignment data, which can be valuable visualizations.)
- Install a TeX distribution with
pdflatex. (e.g. MiKTeX, Tex Live). (Optional; used to convert multiple sequence alignment TeX files to PDF.) - Install pdftk (version 2.02+). (Optional; used to merge individual PDFs into a single file.) If you are unable to install pdftk, but your system has the pdfunite command line utility, you can install the R package
pdfuniter, which performs a similar function. Withpdfuniter, runepfindwithpdftk = FALSE, pdfunite = TRUE.- as of epitopefindr version 1.1.30 (2020-09-20),
pdftk = FALSE, pdfunite = TRUEis the default behavior. If your machine does not have the underlying pdfunite utility (e.g. macOS), trybrew install popplerand thengem install pdfunite.
- as of epitopefindr version 1.1.30 (2020-09-20),
- epitopefindr 1.1.29 (2020-05-30) updates the DESCRIPTION file to specify sources of Bioconductor and Github packages. If the above installation produces issues during certain package installations, try the following:
if (!requireNamespace("BiocManager")) install.packages("BiocManager")
BiocManager::install(c("Biostrings", "IRanges", "msa", "S4Vectors"))
# Install Github packages
if(!requireNamespace("devtools")) install.packages("devtools")
devtools::install_github("mhahsler/rBLAST")
devtools::install_github("brandonsie/pdfuniter")
devtools::install_github("brandonsie/epitopefindr")- Prepare a list of your peptides of interest using one of the following two methods. Either of these can be fed as the first input parameter to
epfind.- Make a FASTA file with peptide names and sequences.
- Make an
AAStringSetobject of peptides (identifier + sequence) as described in the Biostrings documentation.
- To run a typical
epitopefindrpipeline, try callingepfind:
# Basic call
epfind(<path to .fasta>, <path to output dir>)
# Without pdflatex or pdftk
epfind(<path to .fasta>, <path to output dir>,
pdflatex = FALSE, pdftk = FALSE)
# More stringent e-value threshold
epfind(<path to .fasta>, <path to output dir>, e.thresh = 0.0001)You can try running epfind() with some provided example data:
my_peptides <- epitopefindr::pairwise_viral_hits[1:50]
epitopefindr::epfind(data = my_peptides, output.dir = "my_epf_1/")A brief summary of the functions called by epfind:
pbCycleBLASTcycles through each input peptide to find the overlap of its alignment with other peptides from the input. Nested within a call topbCycleBLASTare calls toepitopeBLAST,indexEpitopes.trimEpitopesperforms a second pass through the identified sequences to tidy alignments.indexGroupscollects trimmed sequences into aligning groupsgroupMSAcreates a multiple sequence alignment motif logo for each groupoutputTablecreates a spreadsheet summarizing identified sequences and epitope groups