-
Notifications
You must be signed in to change notification settings - Fork 27
Commit
This commit does not belong to any branch on this repository, and may belong to a fork outside of the repository.
update readme Thu Aug 16 23:07:55 HKT 2018
- Loading branch information
LUO Ruibang
authored and
LUO Ruibang
committed
Aug 16, 2018
1 parent
a713423
commit a7201d8
Showing
3 changed files
with
68 additions
and
38 deletions.
There are no files selected for viewing
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
|
|
@@ -5,13 +5,6 @@ Email: [email protected] | |
|
|
||
| *** | ||
|
|
||
| ## Installation | ||
| ```shell | ||
| git clone --depth=1 https://github.com/aquaskyline/Clairvoyante.git | ||
| cd Clairvoyante | ||
| curl http://www.bio8.cs.hku.hk/trainedModels.tbz | tar -jxf - | ||
| ``` | ||
|
|
||
| ## Introduction | ||
| Identifying the variants of DNA sequences sensitively and accurately is an important but challenging task in the field of genomics. This task is particularly difficult when dealing with Single Molecule Sequencing, the error rate of which is still tens to hundreds of times higher than Next Generation Sequencing. With the increasing prevalence of Single Molecule Sequencing, an efficient variant caller will not only expedite basic research but also enable various downstream applications. To meet this demand, we developed Clairvoyante, a multi-task five-layer convolutional neural network model for predicting variant type, zygosity, alternative allele and Indel length. On NA12878, Clairvoyante achieved 99.73%, 97.68% and 95.36% accuracy on known variants, and achieved 98.65%, 92.57%, 77.89% F1 score on the whole genome, in Illumina, PacBio, and Oxford Nanopore data, respectively. Training Clairvoyante with a sample and call variant on another shows that Clairvoyante is sample agnostic and general for variant calling. A slim version of Clairvoyante with reduced model parameters produced a much lower F1, suggesting the full model's power in disentangling subtle details in read alignment. Clairvoyante is the first method for Single Molecule Sequencing to finish a whole genome variant calling in two hours on a 28 CPU-core machine, with top-tier accuracy and sensitivity. A toolset was developed to train, utilize and visualize the Clairvoyante model easily, and is publically available here is this repo. | ||
|
|
||
|
|
@@ -22,16 +15,24 @@ Identifying the variants of DNA sequences sensitively and accurately is an impor | |
| 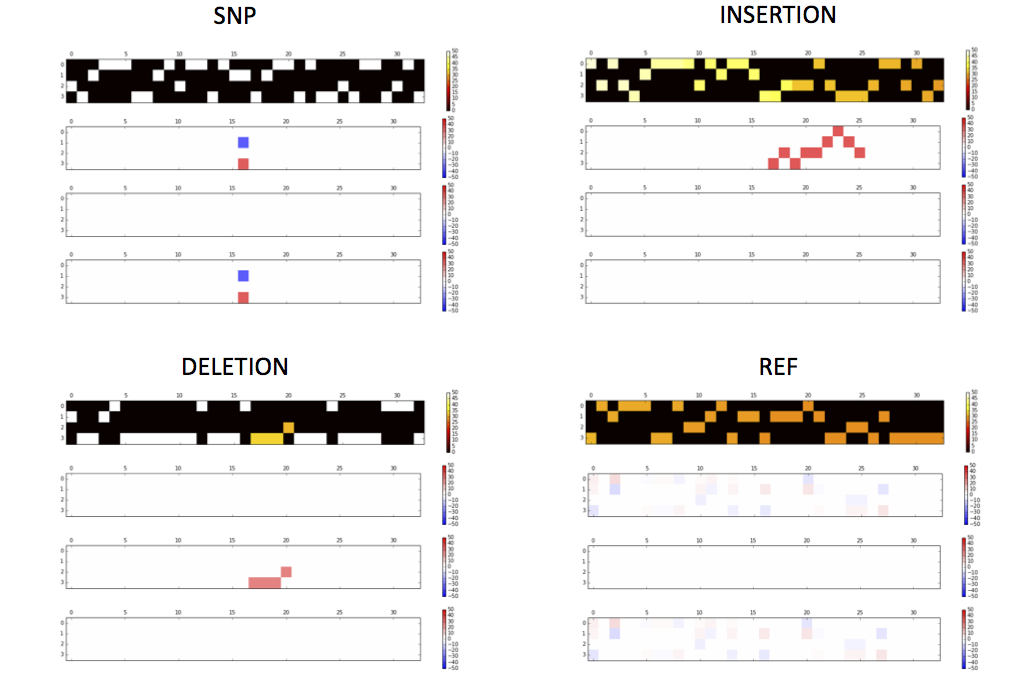 | ||
| ### Activations of the conv1 hidden layer to a non-variant tensor | ||
|  | ||
|
|
||
| *** | ||
| ## Prerequisition | ||
| ### Basics | ||
| Make sure you have Tensorflow ≥ 1.0.0 installed, the following commands install the lastest CPU version of Tensorflow: | ||
|
|
||
| ## Installation | ||
| ### Step by step | ||
| ```shell | ||
| pip install tensorflow | ||
| pip install blosc | ||
| pip install intervaltree | ||
| pip install numpy | ||
| git clone --depth=1 https://github.com/aquaskyline/Clairvoyante.git | ||
| cd Clairvoyante | ||
| curl http://www.bio8.cs.hku.hk/trainedModels.tbz | tar -jxf - | ||
| ``` | ||
|
|
||
| Make sure you have Tensorflow ≥ 1.0.0 installed, the following commands install the lastest CPU version of Tensorflow as well as other dependencies: | ||
|
|
||
| ```shell | ||
| pip install tensorflow | ||
| pip install blosc | ||
| pip install intervaltree | ||
| pip install numpy | ||
| ``` | ||
|
|
||
| To check the version of Tensorflow you have installed: | ||
|
|
@@ -40,27 +41,34 @@ To check the version of Tensorflow you have installed: | |
| python -c 'import tensorflow as tf; print(tf.__version__)' | ||
| ``` | ||
|
|
||
| To do variant calling using trained models, CPU will suffice. Clairvoyante uses all available CPU cores by default in `callVar.py`, use 4 threads by default in `callVarBam.py`, and can be controlled using the parameter `--threads`. To train a new model, a high-end GPU along with the GPU version of Tensorflow is needed. To install the GPU version of tensorflow: | ||
| To do variant calling using trained models, CPU will suffice. Clairvoyante uses all available CPU cores by default in `callVar.py`, 4 threads by default in `callVarBam.py`. The number of threads to be used can be controlled using the parameter `--threads`. To train a new model, a high-end GPU and the GPU version of Tensorflow is needed. To install the GPU version of tensorflow: | ||
|
|
||
| ```shell | ||
| pip install tensorflow-gpu | ||
| ``` | ||
|
|
||
| Clairvoyante was written in Python2 (tested on Python 2.7.10 in Linux and Python 2.7.13 in MacOS). It can be translated to Python3 using "2to3" just like other projects. | ||
|
|
||
| ### Performance of GPUs in model training | ||
| Equiptment | Seconds per Epoch per 11M Variant Tensors | | ||
| :---: |:---:| | ||
| Tesla V100 | 90 | | ||
| GTX 1080 Ti | 170 | | ||
| GTX 980 | 350 | | ||
| GTX Titan | 520 | | ||
| Tesla K40 (-ac 3004,875) | 580 | | ||
| Tesla K40 | 620 | | ||
| Tesla K80 (one socket) | 600 | | ||
| GTX 680 | 780 | | ||
| Intel Xeon E5-2680 v4 28-core | 2900 | ||
| Using pure Python interpreter on Clairvoyante is slow. Please refer to the **Speed up with PyPy** section for speed up. | ||
|
|
||
| ### Using bioconda | ||
|
|
||
| ```shell | ||
| conda create -n clairvoyante-conda-env -c bioconda clairvoyante | ||
| source activate clairvoyante-conda-env | ||
| pypy -m pip install intervaltree | ||
| pypy -m pip install blosc | ||
| ``` | ||
|
|
||
| The commands above install the CPU version of TensorFlow in the virtual environment thus support only variant calling. To train a model, please install the GPU version of Tensorflow in the virtual environment: | ||
|
|
||
| ```shell | ||
| conda remove tensorflow | ||
| conda install tensorflow-gpu | ||
| ``` | ||
|
|
||
| Use `source deactivate` to exit the virtual environment. | ||
| Use `source activate clairvoyante-conda-env` to re-enter the virtual environment. | ||
|
|
||
| ### Speed up with PyPy | ||
| Without a change to the code, using PyPy python interpreter on some tensorflow independent modules such as `dataPrepScripts/ExtractVariantCandidates.py` and `dataPrepScripts/CreateTensor.py` gives a 5-10 times speed up. Pypy python interpreter can be installed by apt-get, yum, Homebrew, MacPorts, etc. If you have no root access to your system, the official website of Pypy provides a portable binary distribution for Linux. Following is a rundown extracted from Pypy's website (pypy-5.8 in this case) on how to install the binaries. | ||
|
|
@@ -74,7 +82,7 @@ cd pypy-5.8-linux_x86_64-portable/bin | |
| # Use pypy as an inplace substitution of python to run the scripts in dataPrepScripts/ | ||
| ``` | ||
|
|
||
| If you can use apt-get or yum in your system, please install both `pypy` and `pypy-dev` packages. And then install the pip for pypy. | ||
| Alternatively, if you can use apt-get or yum in your system, please install both `pypy` and `pypy-dev` packages. And then install the pip for pypy. | ||
|
|
||
| ```shell | ||
| sudo apt-get install pypy pypy-dev | ||
|
|
@@ -84,7 +92,7 @@ sudo pypy -m pip install blosc | |
| sudo pypy -m pip install intervaltree | ||
| ``` | ||
|
|
||
| To guarantee a good user experience, pypy must be installed to run `callVarBam.py` (call variants from BAM), and `callVarBamParallel.py` that generate parallelizable commands to run `callVarBam.py`. | ||
| To guarantee a good user experience (good speed), pypy must be installed to run `callVarBam.py` (call variants from BAM), and `callVarBamParallel.py` that generate parallelizable commands to run `callVarBam.py`. | ||
| Tensorflow is optimized using Cython thus not compatible with `pypy`. For the list of scripts compatible to `pypy`, please refer to the **Folder Stucture and Program Descriptions** section. | ||
| *Pypy is an awesome Python JIT intepreter, you can donate to [the project](https://pypy.org).* | ||
|
|
||
|
|
@@ -252,16 +260,16 @@ The trained models are in the `trainedModels/` folder. | |
|
|
||
| Folder | Tech | Aligner | Ref | Sample | | ||
| --- |:---:|:---:|:---:|:---:| | ||
| `fullv3-illumina-novoalign-hg001+hg002-hg38` | Illumina HiSeq2500<sup>1</sup> | Nonoalign 3.02.07 | hg38 | NA12878+NA24385 | | ||
| `fullv3-illumina-novoalign-hg001-hg38` | Illumina HiSeq2500<sup>1</sup> | Nonoalign 3.02.07 | hg38 | NA12878 | | ||
| `fullv3-illumina-novoalign-hg002-hg38` | Illumina HiSeq2500<sup>1</sup> | Nonoalign 3.02.07 | hg38 | NA24385 | | ||
| `fullv3-pacbio-ngmlr-hg001+hg002+hg003+hg004-hg19` | mainly PacBio P6-C4<sup>2</sup> | NGMLR 0.2.6 | hg19 | NA12878+NA24385+NA24149+NA24143 | | ||
| `fullv3-pacbio-ngmlr-hg001+hg002-hg19` | PacBio P6-C4<sup>2</sup> | NGMLR 0.2.6 | hg19 | NA12878+NA24385 | | ||
| `fullv3-pacbio-ngmlr-hg001-hg19` | PacBio P6-C4<sup>2</sup> | NGMLR 0.2.6 | hg19 | NA12878 | | ||
| `fullv3-pacbio-ngmlr-hg002-hg19` | PacBio P6-C4<sup>2</sup> | NGMLR 0.2.6 | hg19 | NA24385 | | ||
| `fullv3-ont-ngmlr-hg001-hg19` | Oxford Nanopore Minion R9.4<sup>3</sup> | NGMLR 0.2.6 | hg19 | NA12878 | | ||
| `fullv3-illumina-novoalign`<br>`-hg001+hg002-hg38` | Illumina HiSeq2500<sup>1</sup> | Nonoalign 3.02.07 | hg38 | NA12878+NA24385 | | ||
| `fullv3-illumina-novoalign`<br>`-hg001-hg38` | Illumina HiSeq2500<sup>1</sup> | Nonoalign 3.02.07 | hg38 | NA12878 | | ||
| `fullv3-illumina-novoalign`<br>`-hg002-hg38` | Illumina HiSeq2500<sup>1</sup> | Nonoalign 3.02.07 | hg38 | NA24385 | | ||
| `fullv3-pacbio-ngmlr`<br>`-hg001+hg002+hg003+hg004-hg19` | mainly PacBio P6-C4<sup>2</sup> | NGMLR 0.2.6 | hg19 | NA12878+NA24385+NA24149<br>+NA24143 | | ||
| `fullv3-pacbio-ngmlr`<br>`-hg001+hg002-hg19` | PacBio P6-C4<sup>2</sup> | NGMLR 0.2.6 | hg19 | NA12878+NA24385 | | ||
| `fullv3-pacbio-ngmlr`<br>`-hg001-hg19` | PacBio P6-C4<sup>2</sup> | NGMLR 0.2.6 | hg19 | NA12878 | | ||
| `fullv3-pacbio-ngmlr`<br>`-hg002-hg19` | PacBio P6-C4<sup>2</sup> | NGMLR 0.2.6 | hg19 | NA24385 | | ||
| `fullv3-ont-ngmlr`<br>`-hg001-hg19` | Oxford Nanopore Minion R9.4<sup>3</sup> | NGMLR 0.2.6 | hg19 | NA12878 | | ||
|
|
||
| <sup>1</sup> Also using Illumina TruSeq (LT) DNA PCR-Free Sample Prep Kits, *Zook et al. Extensive sequencing of seven human genomes to characterize benchmark reference materials. 2016* | ||
| <sup>1</sup> Also using Illumina TruSeq (LT) DNA PCR-Free Sample Prep Kits. *Zook et al. Extensive sequencing of seven human genomes to characterize benchmark reference materials. 2016* | ||
|
|
||
| <sup>2</sup> *Pendelton et al. Assembly and diploid architecture of an individual human genome via single-molecule technologies. 2015* | ||
|
|
||
|
|
@@ -271,6 +279,21 @@ Folder | Tech | Aligner | Ref | Sample | | |
|
|
||
| *** | ||
|
|
||
| ## Performance of GPUs in model training | ||
| Equiptment | Seconds per Epoch per 11M Variant Tensors | | ||
| :---: |:---:| | ||
| Tesla V100 | 90 | | ||
| GTX 1080 Ti | 170 | | ||
| GTX 980 | 350 | | ||
| GTX Titan | 520 | | ||
| Tesla K40 (-ac 3004,875) | 580 | | ||
| Tesla K40 | 620 | | ||
| Tesla K80 (one socket) | 600 | | ||
| GTX 680 | 780 | | ||
| Intel Xeon E5-2680 v4 28-core | 2900 | ||
|
|
||
| *** | ||
|
|
||
| ## About Setting the Alternative Allele Frequency Cutoff | ||
|
|
||
| Different from model training, in which all genome positions are candidates but randomly subsampled for training, variant calling using a trained model will require the user to define a minimal alternative allele frequency cutoff for a genome position to be considered as a candidate for variant calling. For all sequencing technologies, the lower the cutoff, the lower the speed. Setting a cutoff too low will increase the false positive rate significantly, while too high will increase the false negative rate significantly. The option `--threshold` controls the cutoff in these three scripts `callVarBam.py`, `callVarBamParallel.py` and `ExtractVariantCandidates.py`. The suggested cutoff is listed below for different sequencing technologies. A higher cutoff will increase the accuracy of datasets with poor sequencing quality, while a lower cutoff will increase the sensitivity in applications like clinical research. Setting a lower cutoff and further filter the variants by their quality is also a good practice. | ||
|
|
||
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
| @@ -0,0 +1,2 @@ | ||
| blosc | ||
| intervaltree |
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
| @@ -0,0 +1,5 @@ | ||
| #install tensorflow-gpu if you want to train models | ||
| tensorflow | ||
| numpy | ||
| blosc | ||
| intervaltree |