| title | tags | authors | bibliography | date | affiliations | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Annoroad-OMIC-Viz: A biological multi-omic visualization tool |
|
|
paper.bib |
18 October 2021 |
|
Different types of high throughput sequencing such as Hi-C, ChIP-seq, ATAC-seq, Cut&Tag, transcriptome, BS-seq, WGS have revolutionized multi-omic science. In turn, these technologies also generated some challenges, such as how to deal with a large amount of data and how to do the integration analysis of different omics data, and after all, find the correlation between different omic data. Big data visualization thus is very important for researchers to discover useful clues. However, drawing a multi-omic track plot that reaches the demand for publication requirement is a big challenge. Currently, published tools we can use are WashU-Browser,pyGenomeTrack, IGV tools, and so on. So even though these tools can help us visualize omic data, however, these tools have various disadvantages. WashU can only run a few species, IGV can't draw the heatmap, pyGenomeTrack is unfriendly to use. All of these tools can not compare differences between two samples' Hi-C data and epigenome or RNA data. Furthermore, these tools can only draw one chromosome area at the same time. If we have so many interesting areas on different chromosomes, then it will be a huge work to draw these pictures one by one. Thus, we developed Annoroad-OMIC-Viz, a command line-based tool to solve this issue. Annoroad-OMIC-Viz is a powerful and flexible tool that perfectly meets the demand of researchers who wants to do integration, visualization, and analysis of epigenetics,3D genomic, methylome WGS and transcriptome data. Besides, our tools can easily combine different tracks in any order according to user needs and generate a vector graph that fits the requirement for publication. Moreover, the picture we generated is highly reproducible. Except for the heat map, all the other data tracks can change the color. Users don’t need to install our package. It only needs python3.6 or above. Besides, the required packages are pandas, numpy, scipy and matplotlib. Among 3D genomics, there are different kinds of data we can perform such as contact matrix, AB-compartment, delta z score matrix between two samples, insulating score, TAD boundary, loops, 4C display. In addition, we can display different kinds of epigenome and transcriptome data, and calculate the difference between two data then show on the track. We support show deletion, duplication, inversion, and insertion from WGS on the track. Based on this handy and powerful tool, we can face the challenge of multi-omic data analysis. For example, deletion may lead to TAD fusion, and TAD fusion can trigger enhancer hijacking and thus cause gene expression alteration. You can use Annoroad-OMIC-Viz to draw all the pictures around deletion which is overlap with the TAD boundary and longer than 10kb. Tracks can include structure variation, heatmaps of two samples, delta Hi-C, epigenome data such as H3K2ac. Based on these data, we can easily find the correlation between structural variation and the 3D structure of chromosomes. Furthermore, with the help of epigenome data, We can identify three-dimensional structural changes caused by structural variations, which in turn lead to changes in gene expression. Here are some examples below, BxPC3, PANC1 are cancer cell lines, HPDE6C7 as the control. We used three examples to illustrate what Annoraod Browser can do.
The left picture first shows three samples' chromosome interaction heatmap, top two are from the cancer cell line, We can see deletions compared with control at the bottom. Below the heatmap, is the 4C display of the MIR31HG of three samples which are distinguished by different colors of line. The x-axis is chromosome position, y-axis represents normalized interaction frequency with MIR31HG promoter. MIR31HG in BxPC3 was deleted, so we can't see the data of BxPC3. PANC1 however, has much stronger interaction at the opposite position of deletion compared with control. At the same time, we can see there are also several peaks on another side of deletion. Thus, we can see that deletion lead to TAD fusion and enhancer hijacking was formed.
The picture in the middle shows the BxPC3 interaction heatmap, below is the structure variation data. Compared with control, BxPC3 has a specific deletion and this deletion leads to TAD fusion. TAD fusion can be further identified by delta Hi-C. This picture can further prove that influence from structure variation was mainly restricted by TAD. So, TAD not only regulates gene expression but also protects the genome structure integrity.
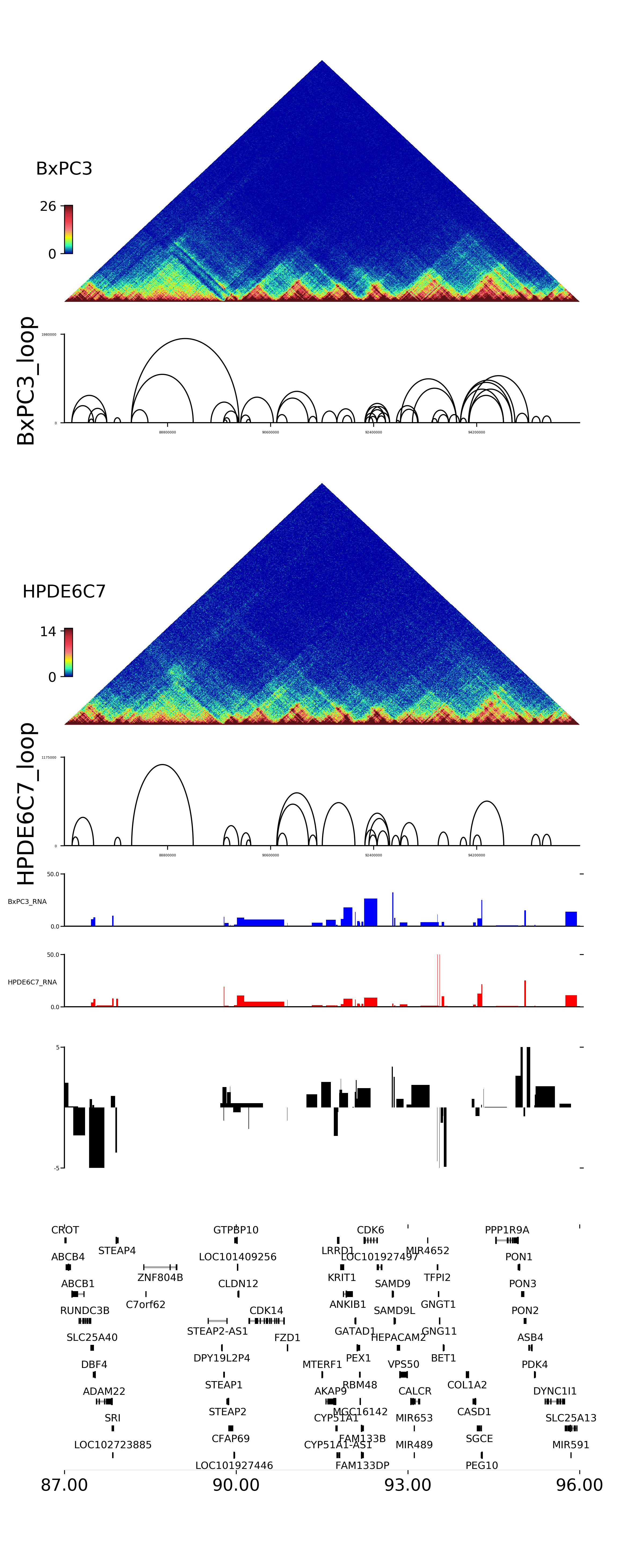
On the right panel, at each sample's heatmap below, are the correspondence loop data. Different samples' TAD structures usually are conserved, however, their loops are highly variable. Below these data, We can see two samples' expression data and its log2 fold change of two samples' gene expression data at the bottom. We can use this kind of joint display to identify the relationship between loop variation and changes in gene expression.


Annoroad Browser is a python3 based tool. It can perform multi-omic visualization. It can display, Hi-C heatmaps. delta-HiC of two samples, loops, AB compartment, TAD boundary, structure variation, epigenomic data, gene track, gene expression and log2 fold change of two samples. Besides, users can also flexibly determine the display sequence of their omic data.
Data that Annoroad Browser can show:
-
Contact matrix: 3D genome, especially Hi-C data is a N by N matrix, this matrix can be raw matrix or ice normalized matrix -
Zscore Delta Matrix: Some times we want to compare the difference of two matrix, this funciont can first do the zscore of two matrices and then do the subtraction -
Gene track: This function can draw the genes on its location of the chromosome which include their exons and introns -
Bedgraph: (Epigenomic data or transcriptome data) epigenome or transcriptome data can be displayed by this function -
Loop: many softwares such as HiCCUPs can find out the loop from chromomse interaction matrix. We can show the loops on the chromosome. -
TAD boundary: chromsome was composed by many different TADs(topological associated domain), thus we want to use this function to perform the TAD boundary -
Structure Variation: structurue variation include deletion, duplication, inservion, inversion. Sometimes structure variation can trigger the 3D alteration of chromosome. This function can show each type's structure varation's postion and thus, we can find relationship between 3D structure and structure variation. -
A/B compartment: A/B compartment is a very import concept of 3D genome. Chromosme usually have two different kinds of compartment, A compartment usually have higher gene expresion level. B compartment have lower gene expression level. -
4C show: 4C means one to many. If you only interest in one postion and want to know how this position interact with other areas, then you can use this function to find out. -
Signal compare: calculate the log2 fold change of two sample's epigenome data or transcriptome data.
Annoroad Browser can display different omic data in one picture, thus we can easier to figure out the association between different omic data. Detailed instructions can found in https://github.com/Spartanzhao/Annoroad-Browser/.
I want to Acknowledge my colleagues in Annoroad Genomics, Fan Xuning helped me to improve the speed of heatmap drawing, Yuan Zan made my heatmap's color scheme more beautiful than before. I also want appreciate the dedicated people of these python packages which include pandas, matplotlib, numpy, scipy.