Multi omic visualization
Multi-omic visualization
Different types of high throughput sequencing such as Hi-C, ChIP-seq, ATAC-seq, Cut&Tag, transcriptome, BS-seq, WGS have revolutionized multi-omic science. In turn, these technologies also generated some challenges, such as how to deal with large amount of data and what how to do the integration analysis of different omic data, and after all, find correlation between different omic data. Big data visualization thus is very important for researchers to discover useful clues. However, draw a multi-omic track plot which reach the demand for publication requirement is a big challange. Thus, we developed Annoroad-OMIC-Viz, a command line based tool to solve this issue. Annoroad-OMIC-Viz is a powerful and flexible tool which perfectly meets the demand of researchers who wants to do integration, visualization and analysis of epigenetic ,3D genomic, methylome WGS and transcriptome data. Besides, out tools can easily combine different tracks in any order according to user needs and generate a vector graph which fits the requirement for publication. Moreover, the picture we generated are highly reproducible. Except heat map, all the other data tracks can change the color. Users doesn’t need to install our package. It only need python3.6 or above. Besides, the requirement packages are pandas, numpy, scipy and matplotlib.
Among 3D genomics, there are different kinds of data we can perform such as contact matrix, A/B compartment, delta z score matrix between two samples, insulating score, TAD boundary, loops, 4C display. In addition, we can display different kinds of epigenome and transcriptome data, and calculate difference between two data then show on the track. We support to show deletion, duplication, inversion and insertion from WGS on the track. Based on this handy and powerful tool, we can face the challenge in multi-omic data analysis. For example, if we draw a track plot, some place on chromosome has a deletion, then the heat map support there is a TAD fusion, another sample have no such deletion and thus have no TAD fusion. On the track of H3K27ac, we find there is a peak in one side of deletion, and after TAD fusion, H3K27ac peak has a strong interaction with a gene on the other side of deletion which formed enhancer hijacting. But the other sample have on such phenomenon. We can further identify it by using delta zscore heatmap in our tool. The transcriptome data showed gene expression is dramatically enhanced after enhancer hijacting. Thus based on our tool, we can easily find such interesting phenomenon very quickly.
This package now can be installed: install method: using anaconda to install dependent packages: python >= 3.6 dependency: argparse>=1.1 re>=2.2.1 numpy>=1.17.3 pandas>=1.0.5 matplotlib>=3.1.1 seaborn>=0.11.0 iced>=0.5.10 scipy>=1.4.1 statsmodels>=0.8.0 for examples: conda install pandas=1.0.5 -y
before you us pip install to install my package, please install these packages as I mentioned above by using Anaconda: such as conda install numpy=1.17.3 -y
Then use command line: pip install Annoroad_OMIC_vis.
After that, you can run: Annoroad_vis -h, If you get : usage: Annoroad_vis [-h] -fl FILE_LIST [-u UPPERCENT] [-d DOWNPERCENT] -o OUTPUT -chr CHROMOSOME -st START -ed END [-t GENETYPE] [-b1 BOUND1] [-b2 BOUND2]
optional arguments: -h, --help show this help message and exit -fl FILE_LIST, --file_list FILE_LIST -u UPPERCENT, --uppercent UPPERCENT up_percent : 85 -d DOWNPERCENT, --downpercent DOWNPERCENT down_percent :5 -o OUTPUT, --output OUTPUT -chr CHROMOSOME, --chrom CHROMOSOME -st START, --start START -ed END, --end END -t GENETYPE, --genetype GENETYPE gene type -b1 BOUND1, --bound1 BOUND1 bound1 -b2 BOUND2, --bound2 BOUND2 bound2
Then that means you have installed this package. Otherwise, please contact me : [email protected]
Data that Annoroad-OMIC-Viz can show:
-
Contact matrix
-
Zscore Delta Matrix
-
Gene track
-
Bedgraph (Epigenomic data or trancriptome data)
-
Loop
-
TAD boundary
-
Structure Variation
-
A/B compartment
-
4C show
10.Signal compare
We need a config file to tell Annoroad-OMIC-Viz what to show. Each row is the track we want to add, different columns was separated by tab. If we want to draw:
1.Contact matrix: The first columns we should write ‘matrix’ which means we want to draw contact matrix. Second column is sample’s name. Third columns is matrix resolution we are going to draw. Fourth columns is matrix list file[1]
-
Delta HiC: The first column should write ‘delta’, the second columns is two sample’s name, separated by comma, third columns is the resolution of two matrix, Fourth columns is matrix list file[1]. The data we show will be first sample’s zscore matrix subtract second sample’s zscore matrix.
-
Gene Track: We shall show the genes in the track, First columns is gtf, the second column is gtf file path.
4 Bedgraph (Epigenomic data or trancriptome data): First column write ‘bedgraph’, second columns is sample’s name, third column is color we choose, fourth columns is data[2], fifth column is maximum number of the y axis. Sixth columns is minimum number of the y axis.
-
Loop: First column write ‘pairwise’, second column is sample’s name, third column is the color we choose. Fourth column is data we choose[3].
-
TAD boundary:
-
Structure Variation First column write ‘SV’, second column is y label of the track. Third column is color, fourth column is SV type, fifth column is structure variation bed file[5]
-
A/B compartment First column write ‘AB’, second column is bin size. Third column is samples name. Fourth column is A/B compartment data[6].
-
4C show First column write ‘4C’, second column is matrix resolution, third column is samples’ name, separated by comma, fourth column is each sample’s color separated by comma, fifth column is observed expected matrix list[4], sixth column is maximum number of y axis.
10 Signal compare First column write ‘delta epi’, second column is the row number of the data we are going to compare in this config file. Third column is another row number of the data we are going to compare in this config file. Fouth column is the maximum number of y axis, fifth column is the minimum number of y axis. The result we showed on the track will be log2(second column/third columns).
[1] format of matrix list: There are three columns: (1) sample name,(2)chromosome name,(3) matrix data{1}
[2] bedgraph data format: There are four columns in the data:(1) chromosome,(2)start,(3)end,(4)signaling
[3] loop data format: There are five columns in the data:(1) chromosome,(2) position of left end of the loop anchor, (3) position of right end of the loop anchor, (4) observed reads who support there are interactions, (5) expected reads who support there are interactions. (if your data have no such information, then you can just use some number to fill the column)
[5] SV data format:
[6] There are three columns: (1) sample name,(2)chromosome name,(3) matrix data{2}
{1} there are three columns:(1) start position of bin,(2) start position of another bin,(3) reads which support these two bins interact with each other.(you can use software HiCCUPs to transform the .hic file into three columns matrix data, then you can use ice method to normalize the matrix if you need)
{2} there are three columns:(1) start position of bin,(2) start position of another bin,(3) reads which support these two bins interact with each other(observed) divide the expected reads which support two bins interaction(expected). (observed/expected matrix can be yield by HiCCUPs)
How to run Annoroad-OMIC-Viz? python3.6 bin/Annoroad_Lego_Browser.py -fl show.list -o MLLT3_4C.png -chr chr9 -st 20620542 -ed 20625542 -b1 1000000 -b2 15000000
-fl: show list file which give the information that you want to show, as described above.
-o: output file
-chr: which chromosome we want to show
-st: start site we want to show.
-ed: end site we want to show.
-b1: left extension,
-b2: right extension, if st is 3000000 ed is 5000000, b1 is 1000000,b2 is 1500000 then we will show the area of from 2000000bp to 6500000bp of this chromosome.
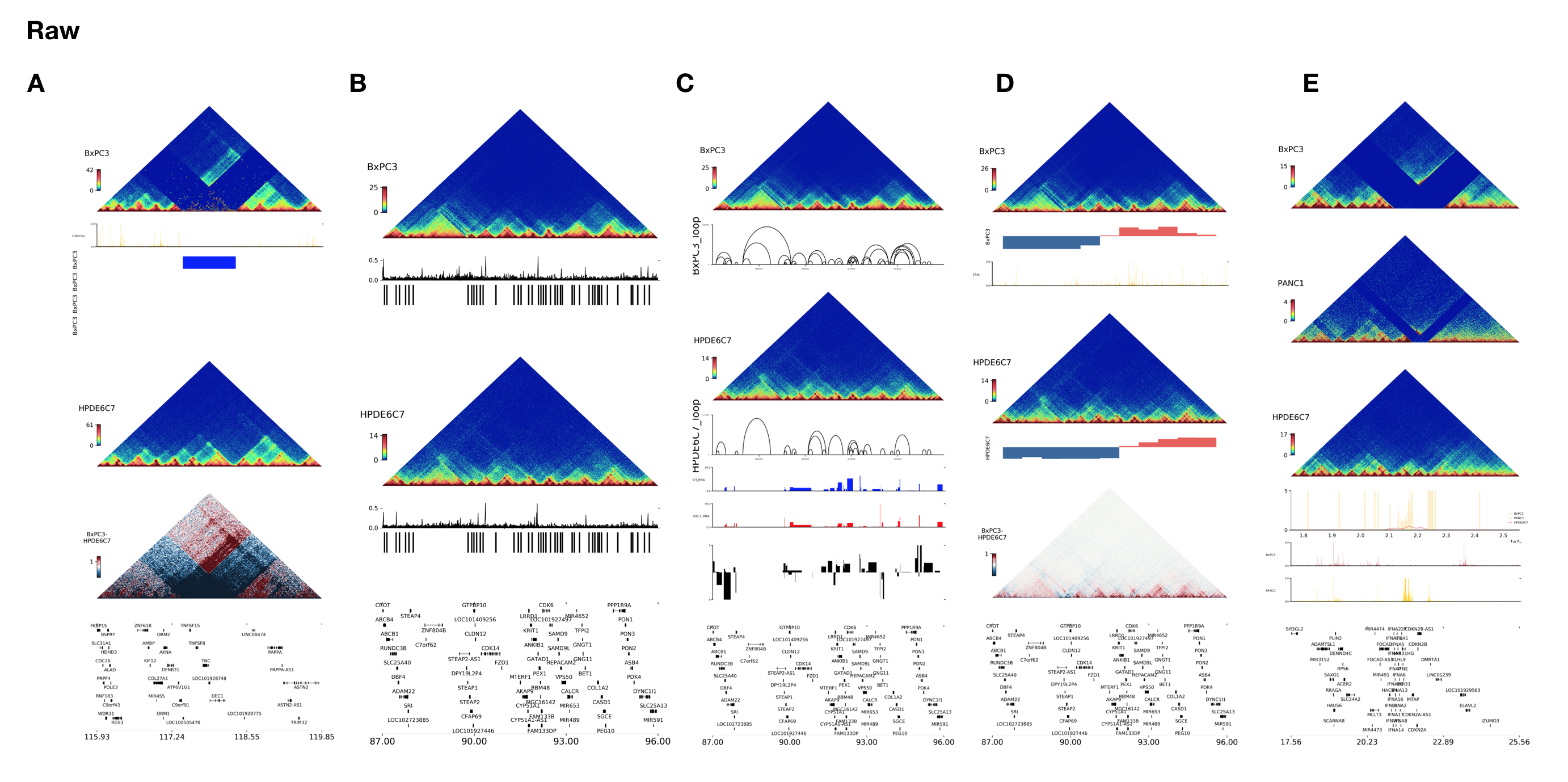
Here are some examples below: We draw 5 different pictures from A to E, and 5 corresponding input files of the Annoroad-OMIC-Viz need are listed above. In figureA, the top one is the Hi-C interaction heatmap of BxPC3, below are BxPC3 H3K27ac ChIP data and SV data. Compared with HPDE6C7 below, interaction between two sides of deletion was dramatically enhanced and there is a potential enhancer hijiackting for we can see that one enhancer on one side of deletion is strongly interacted with a gene on the other side of deletion. The differential interaction we can identfied though delta Hi-C below the SV data. The bottom is gene track.
In figureB, from top to bottom are BxPC3 Hi-C interaction heatmap, BxPC3 CDB LRI score, BxPC3 TAD boundary, HPDE6C7 interaction heatmap, HPDE6C7 CDB LRI score,HPDE6C7 contact domain boundary. The bottom is gene track.
FigureC from top to bottom are BxPC3 Hi-C interaction heatmap, BxPC3 loop, HPDE6C7 Hi-C interaction heatmap, HPDE6C7 loop, BxPC3 gene expression, HPDE6C7 gene expression, Log2FC of BxPC3/HPDE6C7. The bottom is gene track.
FigureD from top to bottom are BxPC3 Hi-C interaction heatmap, BxPC3 A/B Compartment, BxPC3 H3K27ac ChIP data, HPDE6C7 Hi-C interaction heatmap, HPDE6C7 A/B Compartment, Delta Hi-C of BxPC3-HPDE6C7. The bottom is gene track.
FigureE from top to bottom are Hi-C interaction heatmaps of BxPC3, PANC1, HPDE6C7, 4C display of three samples in between start and end site. BxPC3 and PANC1 H3K27ac ChIP data, The bottom is gene track.
Hi-C heatmap data matrix list and oe matrix list are listed in Annoroad-Browser/data/ folder. The matrix data please check Annoroad-Browser/data/required_data.txt. The rest data are in Annoroad-Browser/data/data_test/ folder.
 ,
,

more detailed example is in test folder of this repository, explanation is in paper.md. You can also make your pipeline, example is in bin folder.