Following Phillip E. C. Compeau, Pavel A. Pevzner, and Glenn Tesler's paper: "Why are de Bruijn graphs useful for genome assembly?" and John Hopkins University's notes on de Bruijn graphs, and other research so that I can understand its importance in genome assembly.
Read Compeau et. al's paper for more info: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5531759/.
My knowledge on this is still very limited so bear with me here.
An edge-centric de Bruijn graph is a cyclic Eulerian and Hamiltonian graph in which there exists n^k nodes (where n is the set of symbols;
in nucleotide sequences, this will be 2)
which consists of every length-k substrings, k-mers assigned to each edge and a (k-1)-mer assigned to each node.
Finding a Hamiltonian cycle in our overlap graph is NP-complete, so to avoid solving problems of NP-completeness, we can alternatively observe an algorithmic approach to finding Eulerian cycles instead, which are solvable in linear time. De Bruijn graphs are nice here.
A k-mer is a sequence/substring within our superstring defined by length k.
(Note: Each (k-1)-mer should be unique; the sequence should appear only once.)
For example, given the sequence ATGGCGTGCA, with k-mer length of 3, our 3-mers are:
ATG
TGG
GGC
GCG
CGT
GTG
TGC
GCA
CAA
AAT
(Which we can represent with E = { e(ATG), e(TGG), e(GGC), e(GCG), e(CGT), e(GTG), e(TGC), e(GCA), e(CAA), e(AAT) }, where e is an Edge
object referencing a k-mer sequence in our code, assigning these weights to each respective edge in edges E)
and our (k-1)-mers are V = { v(AT), v(TG), v(GG), v(GC), v(CG), v(GT), v(CA), v(AA) }, where v is a Node object.
If an overlap exists between 2 nodes in our reads by k - 2, we have a k-mer and we can connect those nodes together with an edge!
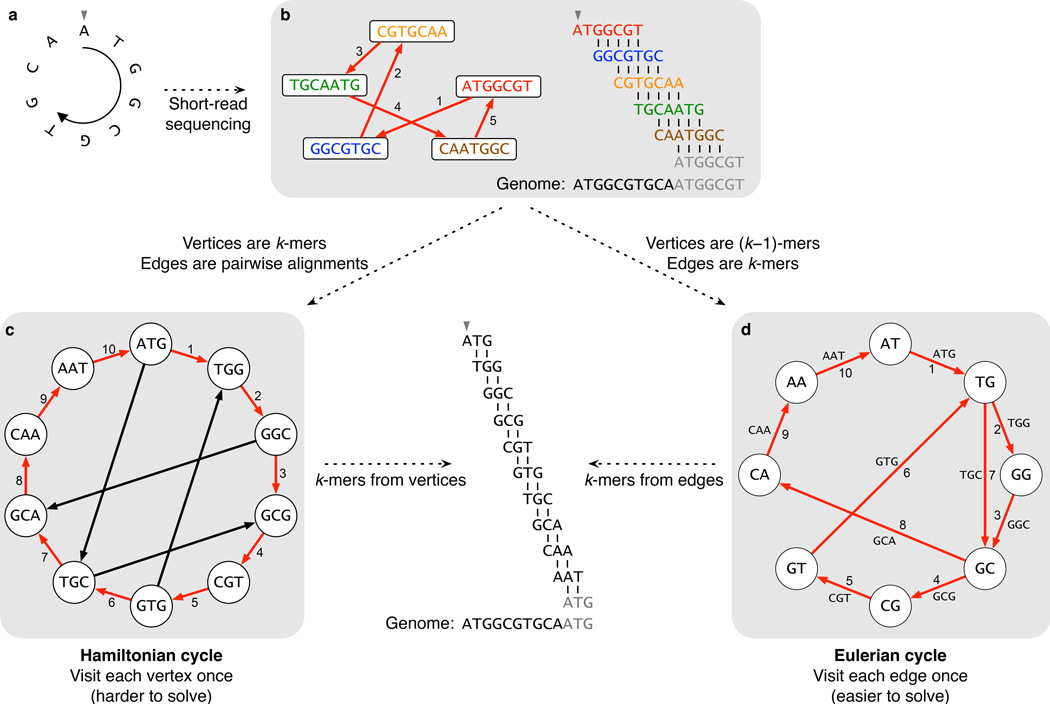
Pulled from Why are de Bruijn graphs useful for genome assembly?
So why construct de Bruijn graphs? In the world of bioinformatics, the construction of de Bruijn graphs are important techniques to assess genome assembly.
As Compeau, Pevzner and Tesler have stated: "A straightforward method for assembling reads into longer contiguous sequences ... uses a graph in which each read is represented by a node and overlap between reads is represented by an arrow (called a directed edge) joining two reads."
Construction of de Bruijn graphs is very popularly used in de novo assembly, in which we create contigs from scratch, and understanding the properties of dBGs can allow us to analyze the computation behind assembling sequences together.
Testing above stated sequence:
ATGGCGTGCA
