Algorithm for Pairwise IBS IBD computation ( genome)
4. Algorithmes
-
Description: Calcule la distance IBS/IBD entre les individus (permet de trouver les liens de parenté).
-
Input: Les fichiers crées avec la commande --make-bed (.bed, .bim et .fam).
-
Output: Le fichier .genome
-
Référence:
Chaque champs du fichier .genome est décrit succinctement ci-dessous, plus de détails seront fournis pour chacun des champs dans la section suivante.
- FID1: Family ID for first sample
- IID1: Individual ID for first sample
- FID2: Family ID for second sample
- IID2: Individual ID for second sample
- RT: Relationship type inferred from .fam/.ped file
- EZ: IBD sharing expected value, based on just .fam/.ped relationship
- Z0: P(IBD=0)
- Z1: P(IBD=1)
- Z2: P(IBD=2)
- PI_HAT: Proportion IBD, i.e. P(IBD=2) + 0.5*P(IBD=1)
- PHE: Pairwise phenotypic code (1, 0, -1 = case-case, case-ctrl, and ctrl-ctrl pairs, respectively)
- DST: IBS distance, i.e. (IBS2 + 0.5*IBS1) / (IBS0 + IBS1 + IBS2)
- PPC: IBS binomial test
- RATIO HETHET: IBS0 SNP ratio (expected value 2)
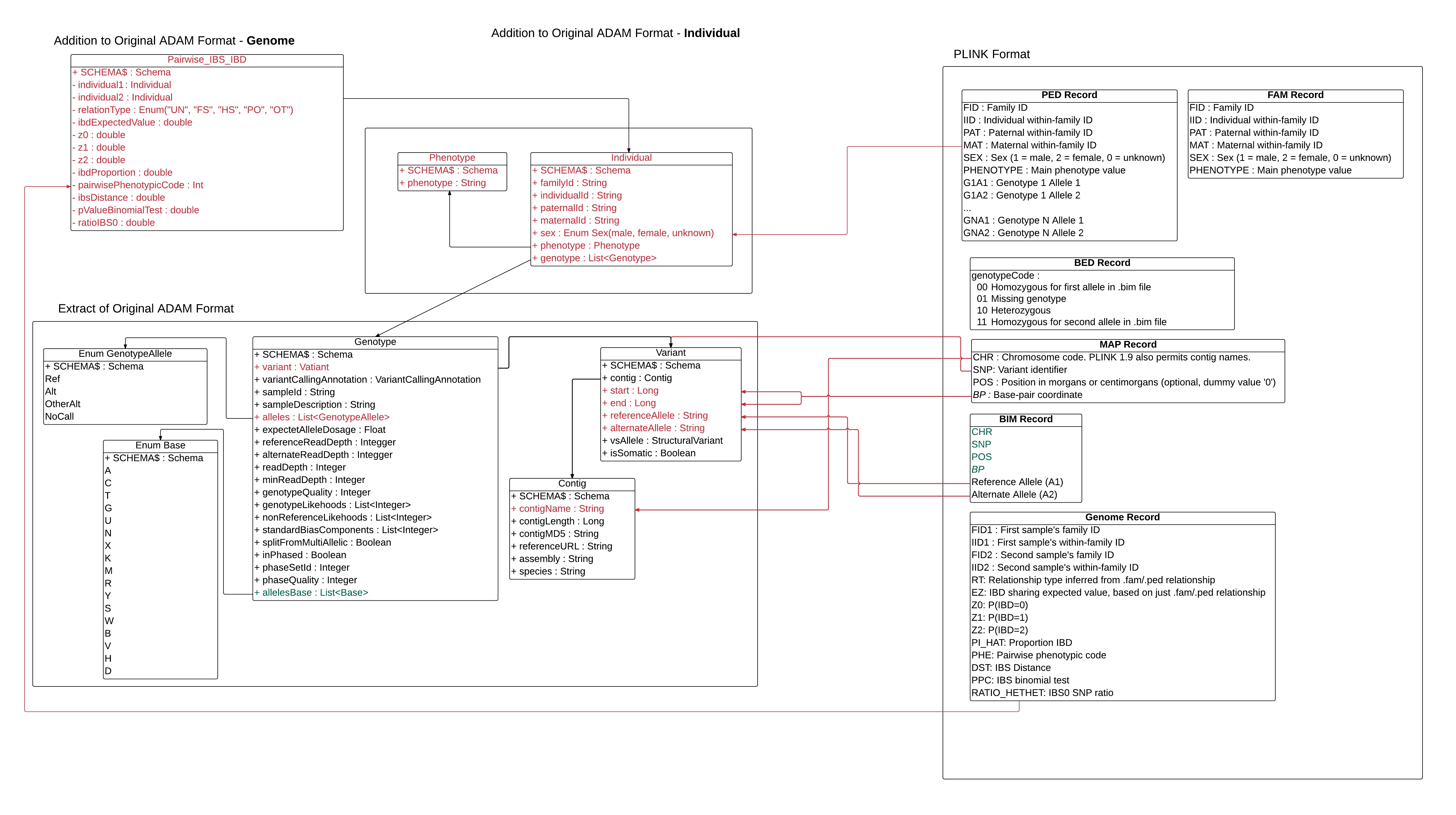
Les 4 premiers champs (FID1, IID1, FID2 et IID2) sont récupérés de la classe Individual qui possède l'ensemble des informations par rapport à un individu (l'id de sa famille, son propre id, son génome etc.)
Référence dans le code source: fichier helper.cpp, la fonction relType() à la ligne 3468 à 3493.
Description:
Le champ RT est "calculé" en comparant les ID des familles (FID) des deux individus, les ID de leurs parents, vérifies si se sont des "founder" (c'est à dire que se sont des parents), et leurs ID individuels (IID). Cette comparaison va permettre de déterminer si les deux individus sont:
- UN: unrelated
- FS: sibling
- HS: half-sibling
- PO: parent-offspring
- OT: other
Algorithme:
Input: Individial a, Individual b
Note: La classe "Individual" possède l'ensemble des attributs nécessaire pour identifier un individu (FID, IID, IDPat, IDMat etc.)
Output: RT
Si (a.FID != b.FID)
retourner "UN"
Si( !( a.founder || b.founder) )
Si (a.IDPAT == b.IDPAT && a.IDMAT == b.IDMAT)
retourner "FS"
else if(a.IDPAT == b.IDPAT || a.IDMAT == b.IDMAT)
retourner "HS"
Si ( a.IDPAT == b.IID || a.IDMAT == b.IID ||
b.IDPAT == a.IID || b.IDMAT == a.IID )
retourner "PO"
retourner "OT"
Référence dans le code source: fichier cfamily.cpp, la fonction genrel() à la ligne 49
Description:
Le champ EZ est "calculé" en comparant les individus passés en paramètre, et vérifies s'ils sont la même personne, s'ils sont des "founder" ou s'ils viennent de famille différente. Cette comparaison va permettre de déterminer à quel point ces deux individus sont semblables.
Algorithme:
Input: Individial a, Individual b
Note: La classe "Individual" possède l'ensemble des attributs nécessaire pour identifier un individu (FID, IID, IDPat, IDMat etc.)
Output: EZ
// Same person?
Si ( a == b )
retourner 1
// Are both individuals founders or in different families?
Si ( a.fid != b.fid )
retourner 0
Si ( a.founder && b.founder )
retourner 0;
Sinon effectuer l'algorithme indiqué à la ligne: 76-167 pour trouver l'ancêtre commun le plus proche.
Description:
Ces champs permettent de déterminer le nombre d'hétérozygote (deux allèles différents) et le nombre d'allèle homozygote (deux allèles identiques).
Algorithme 1: Calcul de IBSg(z0, z1 et z2): voir la classe genome.cpp, la fonction calcGenomeIBS() à la ligne 560.
Créer un vecteur qui contient: z0, z1 et z2
Créer un compteur pour les loci non-absentes.
Créer un intégrateur pour parcourir chacune des séquences de chaque individu
Boucler sur la première séquence du premier individu
On ne prend pas en compte les marqueurs chromosomiques (manquants) -> haploïde et les marqueurs sexuels
// locus[] est un vecteur de booléen: voir le fichier plink.cpp à la ligne 2127.
if( par::chr_sex[locus[l]] -> chr || par::chr_haploid[locus[l]] -> chr) continue; // voir ligne 588 et 589 de la fonction calcGenomeIBS()
On ne considère que les génotypes présents (voir lignes 599 à 622)
Calculer IBS du génotype (voir le calcul à la ligne 625 à 679)
Note: SNP (Single Nucleotide Phenotype) genotyping: mesure de la variation d’un seul nucléotide (un nucléotide est représenté par les lettres A,T,G,C).
loci autosomique: qualifie un gène situé sur un autosome, c’est à dire un chromosome non sexuel.
Voir la classe plein.h à la ligne 545 pour l’attribut « locus » (vector<Locus*> locus)
Algorithme 2: Calcul des EXY: voir la classe genome.cpp, la fonction preCalcGenomeIBD() à ligne 720
Suivre l’algorithme de la fonction preCalcGenomeIBD()
E00 /= cnt; E10 = 0; E20 = 0;
E01 /= cnt; E11 /= cnt; E21 = 0;
E02 /= cnt; E12 /= cnt; E22 = 1;
_Note: nl_all: tous les loci. nl_all = locus.size() // défini dans plink.h, ligne 566 et initialiser dans le fichier plink.cpp à la ligne 2127. _
Algorithme 3: alcul des Z0, Z1 et Z2, voir la classe genome.cpp, la fonction calcGenomeIBD() à ligne 821.
Note: Cette fonction prend en paramètre le IBSg calculer par la fonction calcGenomeIBS() et les EXY calculés par la fonction preCalcGenomeIBD().
// Calcul du S
S = IBSg.z0 + IBSg.z1 + IBSg.z2
// Calcul des eXY
e00 = E00*S;
e10 = E10*S;
e20 = E20*S;
e01 = E01*S;
e11 = E11*S;
e21 = E21*S;
e02 = E02*S;
e12 = E12*S;
e22 = E22*S;
// Calcul des Z0, Z1 et Z2
Z0 = IBSg.z0 / e00
Z1 = (IBSg.z1 -z0 * 201)/e11
Z2 = (IBSg.z2 - z0 *e02 - z1 * e12)/e22
Vérifier si Z0, Z1 et Z2 font parties de l’intervalle [0-1] (voir ligne 858 à 867)
Référence dans le code source: fichier genome.cpp, la fonction calcGenomeIBD() à la ligne 884
Description:
Le PI_HAT est une proportion IBD basé sur le Z1 et le Z2 calculé précédemment.
La formule est la suivante: PI_HAT = Z2 + 0.5*Z1
Référence dans le code source: fichier genome.cpp, la fonction calcGenomeIBD() à la ligne 951 à 965
Description:
Le champ "PHE" représente le code phénotype de la paire d'individus qui est comparée. La comparaison s'effectue selon l'état de l'individu: s'il est affecté ou non.
Il y a 3 cas à déterminer:
- Case-Case: les deux allèles sont affectés (la valeur retournée est 1).
- Case-Control: l'un des allèles est affectés (la valeur retournée est 0).
- Control-Control: aucun allèle n'est affecté (la valeur retournée est -1).
Algorithme:
Input: Individial a, Individual b
Note:
- pX.aff: Signifie que l'allèle de l’individu est affecté (on l'appelle le "case")
- !pX.aff: Signifie que l'allèle de l’individu n’est pas affecté (on l'appelle le "control")
- par::bt vient de la classe « nested class » par qui se trouve dans la classe Options.h
Output: PHE
Si (par::bt)
Si (!p1->aff) && (!p2->aff)
Afficher « -1 »
Sinon si (p1->aff) && (p2->aff)
Afficher « 1 »
Sinon si (!p1->aff) && (p2->aff)
Afficher « 0 »
Sinon si (p1->aff) && (!p2->aff)
Afficher « 0 »
Sinon
Afficher « NA »
Sinon
Afficher « NA »
Référence dans le code source: fichier genome.cpp, la fonction calcGenomeIBS() à la ligne 707.
Description:
Le champ "DST" fournie l'information sur la distance de la comparaison IBS.
La formule est la suivante: DST = (IBS2 + 0.5*IBS1)/ (IBS0 + IBS1 + IBS2)
Référence dans le code source: Aucune.
Description:
Le champ "PPC" représente le p-value en statistique pour le test binomial.
La valeur du PPC est 0.05 qui est la valeur standard en statistique.
Référence dans le code source: fichier genome.cpp, la fonction calculGenomeIBD(), à la ligne 970.
Description:
Le champ "RATIO HETHET" fait le ratio entre les compteurs "pvIBS2het" et le "pvIBS0".
Note: voir la fonction calcGenomeIBS() de la classe genome.cpp pour connaître les détails des calculs de ces compteurs
ratio ov = pvIBS2het / pvIBS0;
Si ov est un nombre
Afficher ov
Sinon
Afficher « NA »