Home
The following details the steps involved in:
- Generating a Trinity de novo RNA-Seq assembly
- Evaluating the quality of the assembly
- Quantifying transcript expression levels
- Identifying differentially expressed (DE) transcripts
- Functionally annotating transcripts using Trinotate and predicting coding regions using TransDecoder
- Examining functional enrichments for DE transcripts using GOseq
- Interactively Exploring annotations and expression data via TrinotateWeb
All required software and data are provided pre-installed on the Amazon EC2 AMI 'ami-2f6f3445'. Be sure to have a running instance on at least an m3.large instance type, so we'll have adequate computational resources allocated in addition to reasonable response times over the network.
The workshop materials here expect that you have a minimal familiarity with UNIX.
After launching and connecting to your running instance of the AMI, change your working directory to the base directory of these workshop materials. We'll get there by moving down three directories:
% cd workshop_data
% cd transcriptomics
% cd KrumlovTrinityWorkshopJan2016
Yes, you could have just 'cd workshop_data/transcriptomics/KrumlovTrinityWorkshopJan2016', but here we're taking the slower scenic route. Feel free to look around and run 'ls' to see what other files or directories are there along the way and get a feel for where you are in the file system - which is particularly useful if you're new to unix.
This 'KrumlovTrinityWorkshopJan2016' will be the base working directory for all the exercises below. We'll create other subdirectories here and move back and forth to our various workspaces that we generate along the way.
Below, we refer to '%' as the terminal command prompt, and we use environmental variables such as $TRINITY_HOME and $TRINOTATE_HOME as shortcuts to referring to their installation directories in the AMI image. To view the path to the installation directories, you can simply run:
% echo $TRINITY_HOME
Also, some commands can be fairly long to type in, an so they'll be more easily displayed in this document, we separate parts of the command with '' characters and put the rest of the command on the following line. Similarly in unix you can type '[return]' and you can continue to type in the rest of the command on the new line in the terminal.
For viewing text files, we'll use the unix utilities 'head' (look at the top few lines), 'cat' (print the entire contents of the file), and 'less' (interactively page through the file), and for viewing PDF formatted files, we'll use the 'xpdf' viewer utility.
Because the workshop materials have been tweaked ever so slightly after we had built the final AMI, we'll need to update the materials in this workshop data space to capture the latest content.
To update, run the following two commands:
% rm data/mini_sprot.pep
% git pull
Assuming no fatal errors occurred, you should now be ready for running the exercises below.
For this course we will be using the data from this paper: Defining the transcriptomic landscape of Candida glabrata by RNA-Seq. Linde et al. Nucleic Acids Res. 2015 This work provides a detailed RNA-Seq-based analysis of the transcriptomic landscape of C. glabrata in nutrient-rich media (WT), as well as under nitrosative stress (GSNO), in addition to other conditions, but we'll restrict ourselves to just WT and GSNO conditions for demonstration purposes in this workshop.
There are paired-end FASTQ formatted Illlumina read files for each of the two conditions, with three biological replicates for each. All RNA-Seq data sets can be found in the data/ subdirectory:
% ls -1 data | grep fastq
.
GSNO_SRR1582646_1.fastq
GSNO_SRR1582646_2.fastq
GSNO_SRR1582647_1.fastq
GSNO_SRR1582647_2.fastq
GSNO_SRR1582648_1.fastq
GSNO_SRR1582648_2.fastq
wt_SRR1582649_1.fastq
wt_SRR1582649_2.fastq
wt_SRR1582650_1.fastq
wt_SRR1582650_2.fastq
wt_SRR1582651_1.fastq
wt_SRR1582651_2.fastq
Each biological replicate (eg. wt_SRR1582651) contains a pair of fastq files (eg. wt_SRR1582651_1.fastq.gz for the 'left' and wt_SRR1582651_2.fastq.gz for the 'right' read of the paired end sequences). Normally, each file would contain millions of reads, but in order to reduce running times as part of the workshop, each file provided here is restricted to only 10k RNA-Seq reads.
It's generally good to evaluate the quality of your input data using a tool such as FASTQC. Since exploration of FASTQC reports has already been done in a previous section of this workshop, we'll skip doing it again here - and trust that the quality of these reads meet expectations.
Finally, another set of files that you will find in the data include 'mini_sprot.pep*', corresponding to a highly abridged version of the SWISSPROT database, containing only the subset of protein sequences that are needed for use in this workshop. It's provided and used here only to speed up certain operations, such as BLAST searches, which will be performed at several steps in the tutorial below. Of course, in exploring your own RNA-Seq data, you would leverage the full version of SWISSPROT and not this tiny subset used here.
To generate a reference assembly that we can later use for analyzing differential expression, we'll combine the read data sets for the different conditions together into a single target for Trinity assembly. We do this by providing Trinity with a list of all the left and right fastq files to the --left and --right parameters as comma-delimited like so:
% ${TRINITY_HOME}/Trinity --seqType fq \
--left data/wt_SRR1582649_1.fastq,data/wt_SRR1582651_1.fastq,data/wt_SRR1582650_1.fastq,data/GSNO_SRR1582648_1.fastq,data/GSNO_SRR1582646_1.fastq,data/GSNO_SRR1582647_1.fastq \
--right data/wt_SRR1582649_2.fastq,data/wt_SRR1582651_2.fastq,data/wt_SRR1582650_2.fastq,data/GSNO_SRR1582648_2.fastq,data/GSNO_SRR1582646_2.fastq,data/GSNO_SRR1582647_2.fastq \
--CPU 2 --max_memory 2G --min_contig_length 150
Note, if you see a message about not being able to identify the version of Java, please just ignore it.
Running Trinity on this data set may take 10 to 15 minutes. You'll see it progress through the various stages, starting with Jellyfish to generate the k-mer catalog, then followed by Inchworm to assemble 'draft' contigs, Chrysalis to cluster the contigs and build de Bruijn graphs, and finally Butterfly for tracing paths through the graphs and reconstructing the final isoform sequences.
Running a typical Trinity job requires ~1 hour and ~1G RAM per ~1 million PE reads. You'd normally run it on a high-memory machine and let it churn for hours or days.
The assembled transcripts will be found at 'trinity_out_dir/Trinity.fasta'.
Just to look at the top few lines of the assembled transcript fasta file, you can run:
% head trinity_out_dir/Trinity.fasta
and you can see the Fasta-formatted Trinity output:
>TRINITY_DN506_c0_g1_i1 len=171 path=[149:0-170] [-1, 149, -2]
TGAGTATGGTTTTGCCGGTTTGGCTGTTGGTGCAGCTTTGAAGGGCCTAAAGCCAATTGT
TGAATTCATGTCATTCAACTTCTCCATGCAAGCCATTGACCATGTCGTTAACTCGGCAGC
AAAGACACATTATATGTCTGGTGGTACCCAAAAATGTCAAATCGTGTTCAG
>TRINITY_DN512_c0_g1_i1 len=168 path=[291:0-167] [-1, 291, -2]
ATATCAGCATTAGACAAAAGATTGTAAAGGATGGCATTAGGTGGTCGAAGTTTCAGGTCT
AAGAAACAGCAACTAGCATATGACAGGAGTTTTGCAGGCCGGTATCAGAAATTGCTGAGT
AAGAACCCATTCATATTCTTTGGACTCCCGTTTTGTGGAATGGTGGTG
>TRINITY_DN538_c0_g1_i1 len=310 path=[575:0-309] [-1, 575, -2]
GTTTTCCTCTGCGATCAAATCGTCAAACCTTAGACCTAGCTTGCGGTAACCAGAGTACTT
Note, the sequences you see will likely be different, as the order of sequences in the output is not deterministic.
The FASTA sequence header for each of the transcripts contains the identifier for the transcript (eg. 'TRINITY_DN506_c0_g1_i1'), the length of the transcript, and then some information about how the path was reconstructed by the software by traversing nodes within the graph.
It is often the case that multiple isoforms will be reconstructed for the same 'gene'. Here, the 'gene' identifier corresponds to the prefix of the transcript identifier, such as 'TRINITY_DN506_c0_g1', and the different isoforms for that 'gene' will contain different isoform numbers in the suffix of the identifier (eg. TRINITY_DN506_c0_g1_i1 and TRINITY_DN506_c0_g1_i2 would be two different isoform sequences reconstructed for the single gene TRINITY_DN506_c0_g1). It is useful to perform certain downstream analyses, such as differential expression, at both the 'gene' and at the 'isoform' level, as we'll do later below.
There are several ways to quantitatively as well as qualitatively assess the overall quality of the assembly, and we outline many of these methods at our Trinity wiki.
You can count the number of assembled transcripts by using 'grep' to retrieve only the FASTA header lines and piping that output into 'wc' (word count utility) with the '-l' parameter to just count the number of lines.
% grep '>' trinity_out_dir/Trinity.fasta | wc -l
How many were assembled?
It's useful to know how many transcript contigs were assembled, but it's not very informative. The deeper you sequence, the more transcript contigs you will be able to reconstruct. It's not unusual to assemble over a million transcript contigs with very deep data sets and complex transcriptomes, but as you 'll see below (in the section containing the more informative guide to assembly assessment) a fraction of the transcripts generally best represent the input RNA-Seq reads.
Capture some basic statistics about the Trinity assembly:
% $TRINITY_HOME/util/TrinityStats.pl trinity_out_dir/Trinity.fasta
which should generate data like so. Note your numbers may vary slightly, as the assembly results are not deterministic.
################################
## Counts of transcripts, etc.
################################
Total trinity 'genes': 695
Total trinity transcripts: 695
Percent GC: 44.43
########################################
Stats based on ALL transcript contigs:
########################################
Contig N10: 656
Contig N20: 511
Contig N30: 420
Contig N40: 337
Contig N50: 284
Median contig length: 214
Average contig: 274.44
Total assembled bases: 190738
#####################################################
## Stats based on ONLY LONGEST ISOFORM per 'GENE':
#####################################################
Contig N10: 656
Contig N20: 511
Contig N30: 420
Contig N40: 337
Contig N50: 284
Median contig length: 214
Average contig: 274.44
Total assembled bases: 190738
The total number of reconstructed transcripts should match up identically to what we counted earlier with our simple 'grep | wc' command. The total number of 'genes' is also reported - and simply involves counting up the number of unique transcript identifier prefixes (without the _i isoform numbers). When the 'gene' and 'transcript' identifiers differ, it's due to transcripts being reported as alternative isoforms for the same gene. In our tiny example data set, we unfortunately do not reconstruct any alternative isoforms, and note that alternative splicing in this yeast species may be fairly rare. Tackling an insect or mammal transcriptome would be expected to yeild many alternative isoforms.
You'll also see 'Contig N50' values reported. You'll remmeber from the earlier lectures on genome assembly that the 'N50 statistic indicates that at least half of the assembled bases are in contigs of at least that contig length'. We extend the N50 statistic to provide N40, N30, etc. statistics with similar meaning. As the N-value is decreased, the corresponding length will increase.
Most of this is not quantitatively useful, and the values are only reported for historical reasons - it's simply what everyone used to do in the early days of transcriptome assembly. The N50 statistic in RNA-Seq assembly can be easily biased in the following ways:
-
Overzealous reconstruction of long alternatively spliced isoforms: If an assembler tends to generate many different 'versions' of splicing for a gene, such as in a combinatorial way, and those isoforms tend to have long sequence lengths, the N50 value will be skewed towards a higher value.
-
Highly sensitive reconstruction of lowly expressed isoforms: If an assembler is able to reconstruct transcript contigs for those transcirpts that are very lowly expressed, these contigs will tend to be short and numerous, biasing the N50 value towards lower values. As one sequences deeper, there will be more evidence (reads) available to enable reconstruction of these short lowly expressed transcripts, and so deeper sequencing can also provide a downward skew of the N50 value.
We now move into the section containing more meaningful metrics for evaluating your transcriptome assembly.
A high quality transcriptome assembly is expected to have strong representation of the reads input to the assembler. By aligning the RNA-Seq reads back to the transcriptome assembly, we can quantify read representation. In Trinity, we have a script that will align each of the fastq files of the paired read set to the assembly separately, and then link up the pairs. This way, we can count the number of reads that are found as properly paired alignments in addition to those that align to separate contigs (improperly paired), in addition to those cases where only one of the paired reads (the left or the right read of the pair) aligns to a contig. Run the following, which wraps the Bowtie software to do the read alignments:
% $TRINITY_HOME/util/bowtie_PE_separate_then_join.pl \
--target trinity_out_dir/Trinity.fasta \
--seqType fq \
--left data/wt_SRR1582651_1.fastq \
--right data/wt_SRR1582651_2.fastq \
--aligner bowtie -- -p 2 --all --best --strata -m 300
The output files will exist in a bowtie_out/ subdirectory. You can look at the list of files generated like so:
% ls -ltr bowtie_out/
Among the outputs, you should find a name-sorted bam file (where aligned reads are sorted by their name, instead of by their coordinate) called 'bowtie_out/bowtie_out.nameSorted.bam'. Using this file and another script in the Trinity software suite, we can count the number of reads that correspond to the different alignment categories:
% $TRINITY_HOME/util/SAM_nameSorted_to_uniq_count_stats.pl \
bowtie_out/bowtie_out.nameSorted.bam
.
#read_type count pct
improper_pairs 3552 50.40
proper_pairs 2580 36.61
right_only 527 7.48
left_only 388 5.51
Total aligned reads: 7047
Generally, in a high quality assembly, you would expect to see at least ~70% and at least ~70% of the reads to exist as proper pairs. Our tiny read set used here in this workshop does not provide us with a high quality assembly, as only 36% of aligned reads are mapped as proper pairs, and half of aligned reads (50%) are found as 'improper_pairs', indicating that both paired reads align to the assembly, but they align to different transcript contigs - which is usually the sign of a fractured assembly. In this case, deeper sequencing and assembly of more reads would be expected to lead to major improvements here.
Another very useful metric in evaluating your assembly is to assess the number of fully reconstructed coding transcripts. This can be done by performing a BLASTX search of your assembled transcript sequences to a high quality database of protein sequences, such as provided by SWISSPROT. Searching a large protein database using BLASTX can take a while - longer than we want during this workshop, so instead, we'll search the mini-version of SWISSPROT that comes installed in our data/ directory:
% blastx -query trinity_out_dir/Trinity.fasta \
-db data/mini_sprot.pep -out blastx.outfmt6 \
-evalue 1e-20 -num_threads 2 -max_target_seqs 1 -outfmt 6
The above blastx command will have generated an output file 'blastx.outfmt6', storing only the single best matching protein given the E-value threshold of 1e-20. By running another script in the Trinity suite, we can compute the length representation of best matching SWISSPROT matches like so:
% $TRINITY_HOME/util/analyze_blastPlus_topHit_coverage.pl \
blastx.outfmt6 trinity_out_dir/Trinity.fasta \
data/mini_sprot.pep | column -t
.
#hit_pct_cov_bin count_in_bin >bin_below
100 77 77
90 18 95
80 12 107
70 17 124
60 16 140
50 24 164
40 34 198
30 43 241
20 64 305
10 24 329
The above table lists bins of percent length coverage of the best matching protein sequence along with counts of proteins found within that bin. For example, 77 proteins are matched by more than 90% of their length up to 100% of their length. There are 18 matched by more than 80% and up to 90% of their length. The third column provides a running total, indicating that 95 transcripts match more than 80% of their length, and 107 transcripts match more than 70% of their length, etc.
The count of full-length transcripts is going to be dependent on how good the assembly is in addition to the depth of sequencing, but should saturate at higher levels of sequencing. Performing this full-length transcript analysis using assemblies at different read depths and plotting the number of full-length transcripts as a function of sequencing depth will give you an idea of whether or not you've sequenced deeply enough or you should consider doing more RNA-Seq to capture more transcripts and obtain a better (more complete) assembly.
We'll explore some additional metrics that are useful in assessing the assembly quality below, but they require that we estimate expression values for our transcripts, so we'll tackle that first.
To estimate the expression levels of the Trinity-reconstructed transcripts, we use the strategy supported by the RSEM software involving read alignment followed by expectation maximization to assign reads according to maximum likelihood estimates. In essence, we first align the original rna-seq reads back against the Trinity transcripts, then run RSEM to estimate the number of rna-seq fragments that map to each contig. Because the abundance of individual transcripts may significantly differ between samples, the reads from each sample (and each biological replicate) must be examined separately, obtaining sample-specific abundance values.
We include a script to faciliate running of RSEM on Trinity transcript assemblies. The script we execute below will run the Bowtie aligner to align reads to the Trinity transcripts, and RSEM will then evaluate those alignments to estimate expression values. Again, we need to run this separately for each sample and biological replicate (ie. each pair of fastq files).
Let's start with one of the GSNO treatment fastq pairs like so:
% $TRINITY_HOME/util/align_and_estimate_abundance.pl --seqType fq \
--left data/GSNO_SRR1582648_1.fastq \
--right data/GSNO_SRR1582648_2.fastq \
--transcripts trinity_out_dir/Trinity.fasta \
--output_prefix GSNO_SRR1582648 \
--est_method RSEM --aln_method bowtie \
--trinity_mode --prep_reference --coordsort_bam \
--output_dir GSNO_SRR1582648.RSEM
The outputs generated from running the command above will exist in the GSNO_SRR1582648.RSEM/ directory, as we indicate with the --output_dir parameter above.
The primary output generated by RSEM is the file containing the expression values for each of the transcripts. Examine this file like so:
% head GSNO_SRR1582648.RSEM/GSNO_SRR1582648.isoforms.results | column -t
and you should see the top of a tab-delimited file:
transcript_id gene_id length effective_length expected_count TPM FPKM IsoPct
TRINITY_DN0_c0_g1_i1 TRINITY_DN0_c0_g1 328 198.75 29.00 9093.16 43883.19 100.00
TRINITY_DN0_c0_g2_i1 TRINITY_DN0_c0_g2 329 199.75 0.00 0.10 0.48 100.00
TRINITY_DN100_c0_g1_i1 TRINITY_DN100_c0_g1 198 69.79 1.00 892.99 4309.53 100.00
TRINITY_DN101_c0_g1_i1 TRINITY_DN101_c0_g1 233 104.12 2.00 1197.08 5777.04 100.00
TRINITY_DN102_c0_g1_i1 TRINITY_DN102_c0_g1 198 69.79 0.00 0.00 0.00 0.00
TRINITY_DN103_c0_g1_i1 TRINITY_DN103_c0_g1 346 216.72 7.00 2012.95 9714.40 100.00
TRINITY_DN104_c0_g1_i1 TRINITY_DN104_c0_g1 264 134.91 1.00 461.94 2229.29 100.00
TRINITY_DN105_c0_g1_i1 TRINITY_DN105_c0_g1 540 410.62 19.00 2883.65 13916.35 100.00
TRINITY_DN106_c0_g1_i1 TRINITY_DN106_c0_g1 375 245.67 3.00 761.01 3672.58 100.00
The key columns in the above RSEM output are the transcript identifier, the 'expected_count' corresponding to the number of RNA-Seq fragments predicted to be derived from that transcript, and the 'TPM' or 'FPKM' columns, which provide normalized expression values for the expression of that transcript in the sample.
The FPKM expression measurement normalizes read counts according to the length of transcripts from which they are derived (as longer transcripts generate more reads at the same expression level), and normalized according to sequencing depth. The FPKM acronym stand for 'fragments per kilobase of cDNA per million fragments mapped'.
TPM 'transcripts per million' is generally the favored metric, as all TPM values should sum to 1 million, and TPM nicely reflects the relative molar concentration of that transcript in the sample. FPKM values, on the other hand, do not always sum to the same value, and do not have the similar property of inherrently representing a proportion within a sample, making comparisons between samples less straightforward. TPM values can be easily computed from FPKM values like so: TPMi = FPKMi / (sum all FPKM values) * 1 million.
Note, a similar RSEM outputted gene expression file exists for the 'gene' level expression data. For example:
% head GSNO_SRR1582648.RSEM/GSNO_SRR1582648.genes.results | column -t
.
gene_id transcript_id(s) length effective_length expected_count TPM FPKM
TRINITY_DN0_c0_g1 TRINITY_DN0_c0_g1_i1 328.00 198.75 29.00 9093.16 43883.19
TRINITY_DN0_c0_g2 TRINITY_DN0_c0_g2_i1 329.00 199.75 0.00 0.10 0.48
TRINITY_DN100_c0_g1 TRINITY_DN100_c0_g1_i1 198.00 69.79 1.00 892.99 4309.53
TRINITY_DN101_c0_g1 TRINITY_DN101_c0_g1_i1 233.00 104.12 2.00 1197.08 5777.04
TRINITY_DN102_c0_g1 TRINITY_DN102_c0_g1_i1 198.00 69.79 0.00 0.00 0.00
TRINITY_DN103_c0_g1 TRINITY_DN103_c0_g1_i1 346.00 216.72 7.00 2012.95 9714.40
TRINITY_DN104_c0_g1 TRINITY_DN104_c0_g1_i1 264.00 134.91 1.00 461.94 2229.29
TRINITY_DN105_c0_g1 TRINITY_DN105_c0_g1_i1 540.00 410.62 19.00 2883.65 13916.35
TRINITY_DN106_c0_g1 TRINITY_DN106_c0_g1_i1 375.00 245.67 3.00 761.01 3672.58
Running this on all the samples can be montonous, and with many more samples, advanced users would generally write a short script to fully automate this process. We won't be scripting here, but instead just directly execute abundance estimation just as we did above but on the other five pairs of fastq files. We'll examine the outputs of each in turn, as a sanity check and also just for fun.
Process fastq pair 2:
% $TRINITY_HOME/util/align_and_estimate_abundance.pl --seqType fq \
--left data/GSNO_SRR1582646_1.fastq \
--right data/GSNO_SRR1582646_2.fastq \
--transcripts trinity_out_dir/Trinity.fasta \
--output_prefix GSNO_SRR1582646 \
--est_method RSEM --aln_method bowtie \
--trinity_mode --prep_reference --coordsort_bam \
--output_dir GSNO_SRR1582646.RSEM
Process fastq pair 3:
% $TRINITY_HOME/util/align_and_estimate_abundance.pl --seqType fq \
--left data/GSNO_SRR1582647_1.fastq \
--right data/GSNO_SRR1582647_2.fastq \
--transcripts trinity_out_dir/Trinity.fasta \
--output_prefix GSNO_SRR1582647 \
--est_method RSEM --aln_method bowtie --trinity_mode \
--prep_reference --coordsort_bam \
--output_dir GSNO_SRR1582647.RSEM
Now we're done with processing the GSNO-treated biological replicates, and we'll proceed to now run abundance estimations for the WT samples.
Process fastq pair 4:
% $TRINITY_HOME/util/align_and_estimate_abundance.pl --seqType fq \
--left data/wt_SRR1582649_1.fastq \
--right data/wt_SRR1582649_2.fastq \
--transcripts trinity_out_dir/Trinity.fasta \
--output_prefix wt_SRR1582649 \
--est_method RSEM --aln_method bowtie --trinity_mode \
--prep_reference --coordsort_bam \
--output_dir wt_SRR1582649.RSEM
Process fastq pair 5:
% $TRINITY_HOME/util/align_and_estimate_abundance.pl --seqType fq \
--left data/wt_SRR1582651_1.fastq \
--right data/wt_SRR1582651_2.fastq \
--transcripts trinity_out_dir/Trinity.fasta \
--output_prefix wt_SRR1582651 \
--est_method RSEM --aln_method bowtie --trinity_mode \
--prep_reference --coordsort_bam \
--output_dir wt_SRR1582651.RSEM
Process fastq pair 6 (last one!!):
% $TRINITY_HOME/util/align_and_estimate_abundance.pl --seqType fq \
--left data/wt_SRR1582650_1.fastq \
--right data/wt_SRR1582650_2.fastq \
--transcripts trinity_out_dir/Trinity.fasta \
--output_prefix wt_SRR1582650\
--est_method RSEM --aln_method bowtie --trinity_mode \
--prep_reference --coordsort_bam \
--output_dir wt_SRR1582650.RSEM
Now, given the expression estimates for each of the transcripts in each of the samples, we're going to pull together all values into matrices containing transcript IDs in the rows, and sample names in the columns. We'll make two matrices, one containing the estimated counts, and another containing the TPM expression values that are cross-sample normalized using the TMM method. This is all done for you by the following script in Trinity, indicating the method we used for expresssion estimation and providing the list of individual sample abundance estimate files:
% $TRINITY_HOME/util/abundance_estimates_to_matrix.pl --est_method RSEM \
--out_prefix Trinity_trans \
GSNO_SRR1582648.RSEM/GSNO_SRR1582648.isoforms.results \
GSNO_SRR1582646.RSEM/GSNO_SRR1582646.isoforms.results \
GSNO_SRR1582647.RSEM/GSNO_SRR1582647.isoforms.results \
wt_SRR1582649.RSEM/wt_SRR1582649.isoforms.results \
wt_SRR1582651.RSEM/wt_SRR1582651.isoforms.results \
wt_SRR1582650.RSEM/wt_SRR1582650.isoforms.results
You should find a matrix file called 'Trinity_trans.counts.matrix', which contains the counts of RNA-Seq fragments mapped to each transcript. Examine the first few lines of the counts matrix:
% head -n20 Trinity_trans.counts.matrix | column -t
.
GSNO_SRR1582646 GSNO_SRR1582648 GSNO_SRR1582647 wt_SRR1582649 wt_SRR1582650 wt_SRR1582651
TRINITY_DN562_c0_g1_i1 0.00 0.00 0.00 1.00 1.00 2.00
TRINITY_DN355_c0_g1_i1 0.00 0.00 1.00 2.00 0.00 1.00
TRINITY_DN512_c0_g1_i1 0.00 1.00 1.00 0.00 0.00 0.00
TRINITY_DN229_c0_g1_i1 1.00 0.00 0.00 2.00 4.00 2.00
TRINITY_DN252_c0_g1_i1 1.00 1.00 1.00 2.00 3.00 3.00
TRINITY_DN585_c0_g1_i1 0.00 0.00 0.00 2.00 2.00 2.00
TRINITY_DN417_c0_g1_i1 0.00 0.00 0.00 0.00 2.00 0.00
TRINITY_DN453_c0_g1_i1 0.00 0.00 0.00 1.00 2.00 2.00
TRINITY_DN620_c0_g1_i1 3.00 4.00 4.00 3.00 1.00 0.00
TRINITY_DN99_c0_g1_i1 1.00 1.00 0.00 1.00 0.00 2.00
TRINITY_DN300_c0_g1_i1 0.00 1.00 1.00 0.00 1.00 0.00
TRINITY_DN283_c0_g1_i1 0.00 0.00 0.00 3.00 1.00 1.00
TRINITY_DN80_c0_g1_i1 4.00 4.00 11.00 10.00 8.00 6.00
TRINITY_DN460_c0_g1_i1 1.00 0.00 1.00 7.00 1.00 4.00
TRINITY_DN604_c0_g1_i1 1.00 0.00 1.00 0.00 0.00 0.00
TRINITY_DN472_c0_g1_i1 2.00 4.00 2.00 5.00 3.00 2.00
TRINITY_DN621_c0_g1_i1 3.00 2.00 4.00 3.00 2.00 6.00
TRINITY_DN553_c0_g1_i1 1.00 2.00 1.00 1.00 0.00 2.00
TRINITY_DN615_c0_g1_i1 0.00 1.00 1.00 0.00 0.00 2.00
You'll see that the above matrix has integer values representing the number of RNA-Seq paired-end fragments that are estimated to have been derived from that corresponding transcript in each of the samples. Don't be surprised if you see some values that are not exact integers but rather fractional read counts. This happens if there are multiply-mapped reads (such as to common sequence regions of different isoforms), in which case the multiply-mapped reads are fractionally assigned to the corresponding transcripts according to their maximum likelihood.
The counts matrix will be used by edgeR (or other tools in Bioconductor) for statistical analysis and identifying significantly differentially expressed transcripts.
Now take a look at the first few lines of the normalized expression matrix:
% head -n20 Trinity_trans.TMM.EXPR.matrix | column -t
.
GSNO_SRR1582646 GSNO_SRR1582648 GSNO_SRR1582647 wt_SRR1582649 wt_SRR1582650 wt_SRR1582651
TRINITY_DN562_c0_g1_i1 0.000 0.000 0.000 1846.402 1856.485 5781.775
TRINITY_DN355_c0_g1_i1 0.000 0.000 988.094 2064.647 0.000 1438.325
TRINITY_DN512_c0_g1_i1 0.000 942.340 857.337 0.000 0.000 0.000
TRINITY_DN229_c0_g1_i1 1133.767 0.000 0.000 2180.404 4494.880 3070.453
TRINITY_DN252_c0_g1_i1 588.191 618.376 553.033 1192.466 1884.940 2239.189
TRINITY_DN585_c0_g1_i1 0.000 0.000 0.000 1805.632 1876.280 2450.393
TRINITY_DN417_c0_g1_i1 0.000 0.000 0.000 0.000 3815.064 0.000
TRINITY_DN453_c0_g1_i1 0.000 0.000 0.000 963.468 1997.034 2648.649
TRINITY_DN620_c0_g1_i1 815.183 1183.354 1038.497 862.938 307.195 0.000
TRINITY_DN99_c0_g1_i1 549.958 580.382 0.000 560.178 0.000 1385.479
TRINITY_DN300_c0_g1_i1 0.000 1692.066 1582.840 0.000 1633.715 0.000
TRINITY_DN283_c0_g1_i1 0.000 0.000 0.000 2509.538 871.787 1118.658
TRINITY_DN80_c0_g1_i1 517.487 573.320 1372.092 1398.262 1201.176 865.462
TRINITY_DN460_c0_g1_i1 352.570 0.000 335.331 2582.546 392.798 1695.573
TRINITY_DN604_c0_g1_i1 1207.129 0.000 1112.027 0.000 0.000 0.000
TRINITY_DN472_c0_g1_i1 827.918 1773.521 784.819 2147.171 1368.051 1012.131
TRINITY_DN621_c0_g1_i1 552.234 405.177 707.408 592.076 422.986 1256.485
TRINITY_DN553_c0_g1_i1 491.036 1042.954 463.655 504.082 0.000 1222.161
TRINITY_DN615_c0_g1_i1 0.000 989.753 902.430 0.000 0.000 2596.428
These are the normalized expression values, which have been further cross-sample normalized using TMM normalization to adjust for any differences in sample composition. TMM normalization assumes that most transcripts are not differentially expressed, and linearly scales the expression values of samples to better enforce this property. TMM normalization is described in A scaling normalization method for differential expression analysis of RNA-Seq data, Robinson and Oshlack, Genome Biology 2010.
We use the TMM-normalized expression matrix when plotting expression values in heatmaps and other expression analyses.
It's useful to perform certain downstream analyses, such as differential expression, based on 'gene' level features in addition to the transcript (isoform) level features.
The above RSEM quantitation generated 'gene' expression estimates in addition to the 'isoform' estimates. The 'gene' estimates are in output files named '.genes.results' instead of the '.isoforms.results', and the output formats are similar.
You should now be in the '/home/genomics/workshop_data/transcriptomics/KrumlovTrinityWorkshopJan2016' directory. (You can verify this with the 'pwd' command).
Build a 'gene' expression count matrix and TMM-normalized expression matrix like so:
% $TRINITY_HOME/util/abundance_estimates_to_matrix.pl --est_method RSEM \
--out_prefix Trinity_genes \
GSNO_SRR1582648.RSEM/GSNO_SRR1582648.genes.results \
GSNO_SRR1582646.RSEM/GSNO_SRR1582646.genes.results \
GSNO_SRR1582647.RSEM/GSNO_SRR1582647.genes.results \
wt_SRR1582649.RSEM/wt_SRR1582649.genes.results \
wt_SRR1582651.RSEM/wt_SRR1582651.genes.results \
wt_SRR1582650.RSEM/wt_SRR1582650.genes.results
Now, you should find the following matrices in your working directory:
% ls -1 | grep gene | grep matrix
.
Trinity_genes.TMM.EXPR.matrix
Trinity_genes.counts.matrix
We'll use these later as a target of our DE analysis, in addition to our transcript matrices.
Although we outline above several of the reasons for why the contig N50 statistic is not a useful metric of assembly quality, below we describe the use of an alternative statistic - the ExN50 value, which we assert is more useful in assessing the quality of the transcriptome assembly. The ExN50 indicates the N50 contig statistic (as earlier) but restricted to the top most highly expressed transcripts. Compute it like so:
% $TRINITY_HOME/util/misc/contig_ExN50_statistic.pl Trinity_trans.TMM.EXPR.matrix \
trinity_out_dir/Trinity.fasta > ExN50.stats
View the contents of the above output file:
% cat ExN50.stats | column -t
.
#E min_expr E-N50 num_transcripts
E1 28814.395 247 1
E3 28814.395 328 2
E4 18279.394 247 3
E5 18279.394 258 4
E6 17104.132 258 5
E7 16810.592 328 6
E8 13159.558 303 8
E9 12746.397 328 9
E10 12639.852 328 11
....
E35 3377.805 443 74
E36 3377.805 434 78
E37 3377.805 428 82
E38 3377.805 424 86
E39 3377.805 424 90
E40 3377.805 423 94
E41 3270.612 424 98
E42 3270.612 424 103
E43 2592.034 424 107
E44 2592.034 423 112
E45 2592.034 428 117
....
E69 1906.139 404 272
E70 1901.811 396 281
E71 1503.354 388 289
E72 1503.354 384 298
E73 1503.354 380 307
E74 1503.354 376 317
E75 1503.354 375 326
E76 1503.354 370 336
E77 1503.354 370 346
E78 1398.262 370 356
E79 1393.687 370 366
....
E89 948.242 329 483
E90 948.242 328 497
E91 948.242 320 511
E92 948.242 315 525
E93 948.242 313 540
E94 948.242 313 555
E95 948.242 309 571
E96 883.830 308 588
E97 806.939 305 607
E98 806.939 302 627
E99 608.064 297 650
E100 0.021 285 689
The above table indicates the contig N50 value based on the entire transcriptome assembly (E100), which is a small value (285). By restricting the N50 computation to the set of transcripts representing the top most 65% of expression data, we obtain an N50 value of 420.
Try plotting the ExN50 statistics:
% $TRINITY_HOME/util/misc/plot_ExN50_statistic.Rscript ExN50.stats
% xpdf ExN50.stats.plot.pdf
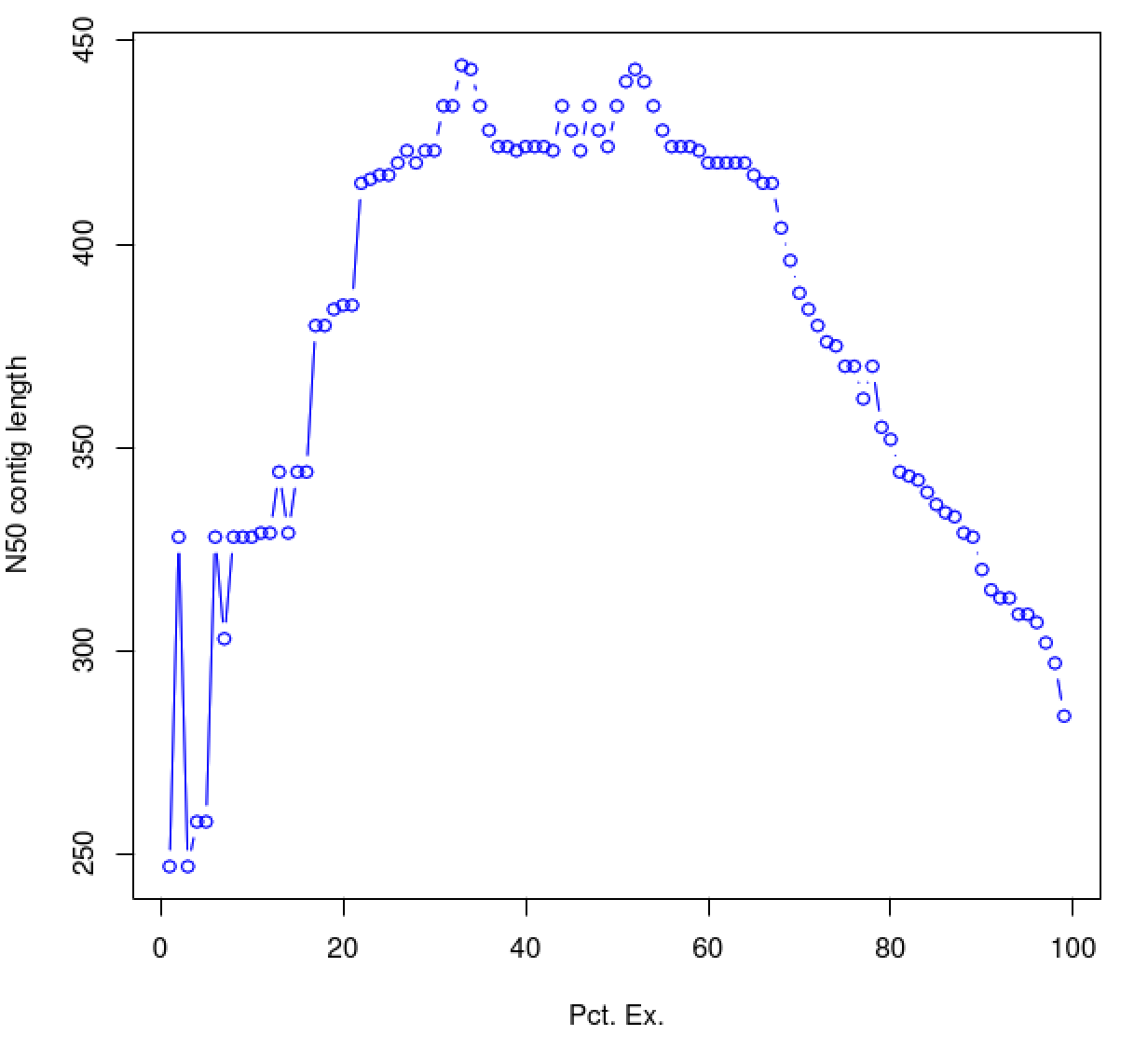
As you can see, the N50 value will tend to peak at a value higher than that computed using the entire data set. With a high quality transcriptome assembly, the N50 value should peak at ~90% of the expression data, which we refer to as the E90N50 value. Reporting the E90N50 contig length and the E90 transcript count are more meaningful than reporting statistics based on the entire set of assembled transcripts. Remember the caveat in assembling this tiny data set. A plot based on a larger set of reads looks like so:
Assembly using 900K PE reads:
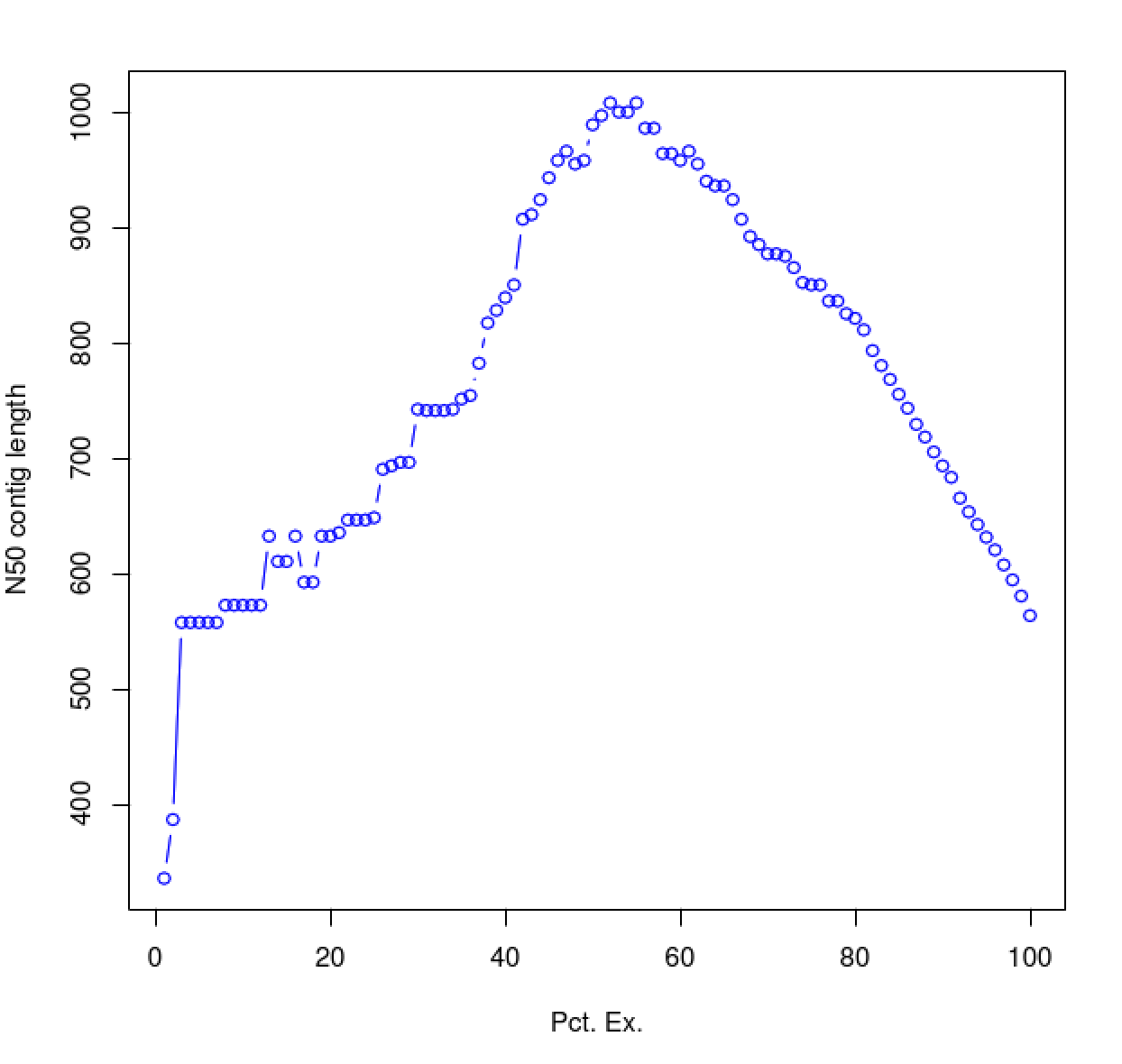
Assembly using 4.5M PE reads:
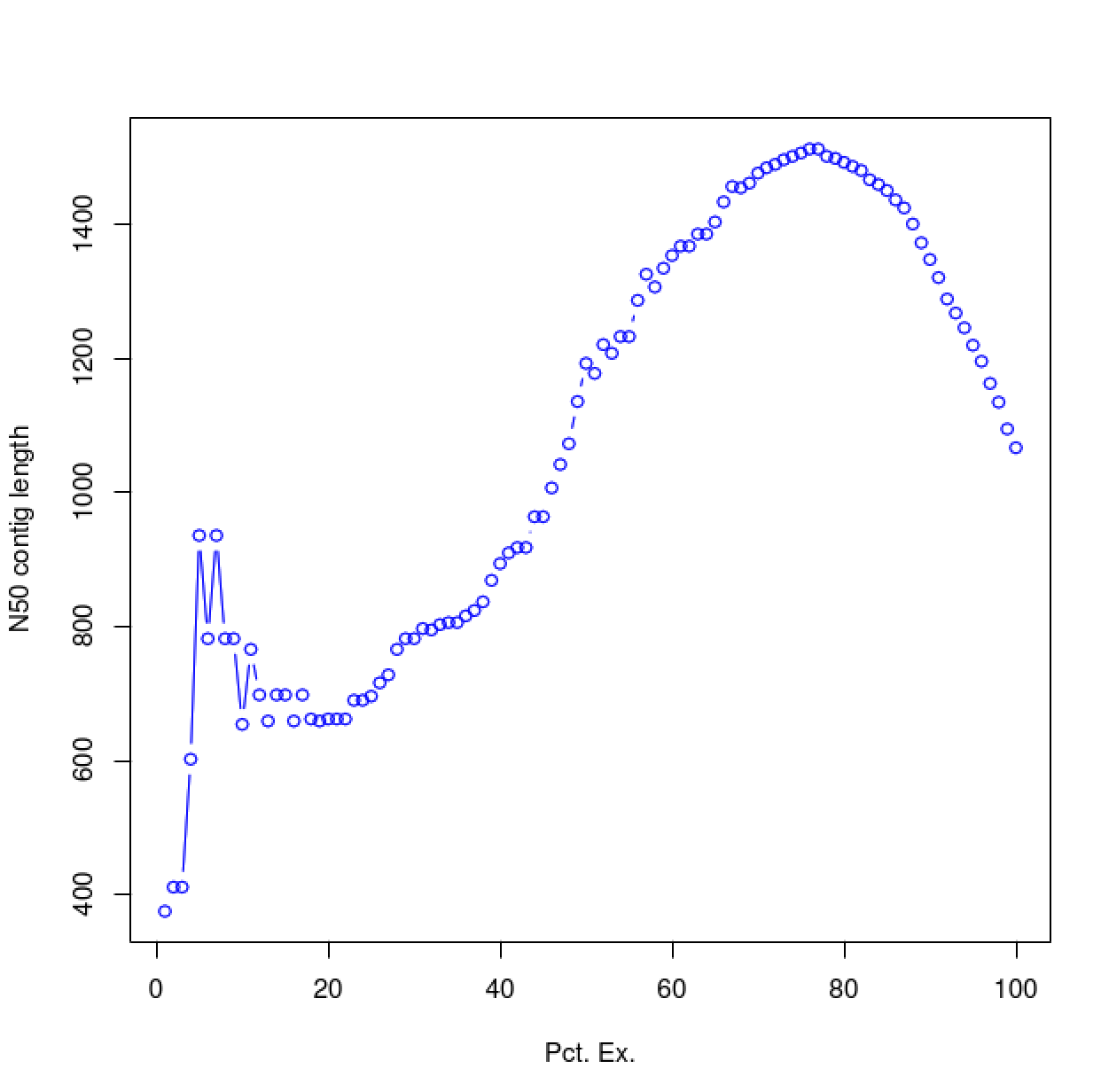
Assembly using 18M PE reads:
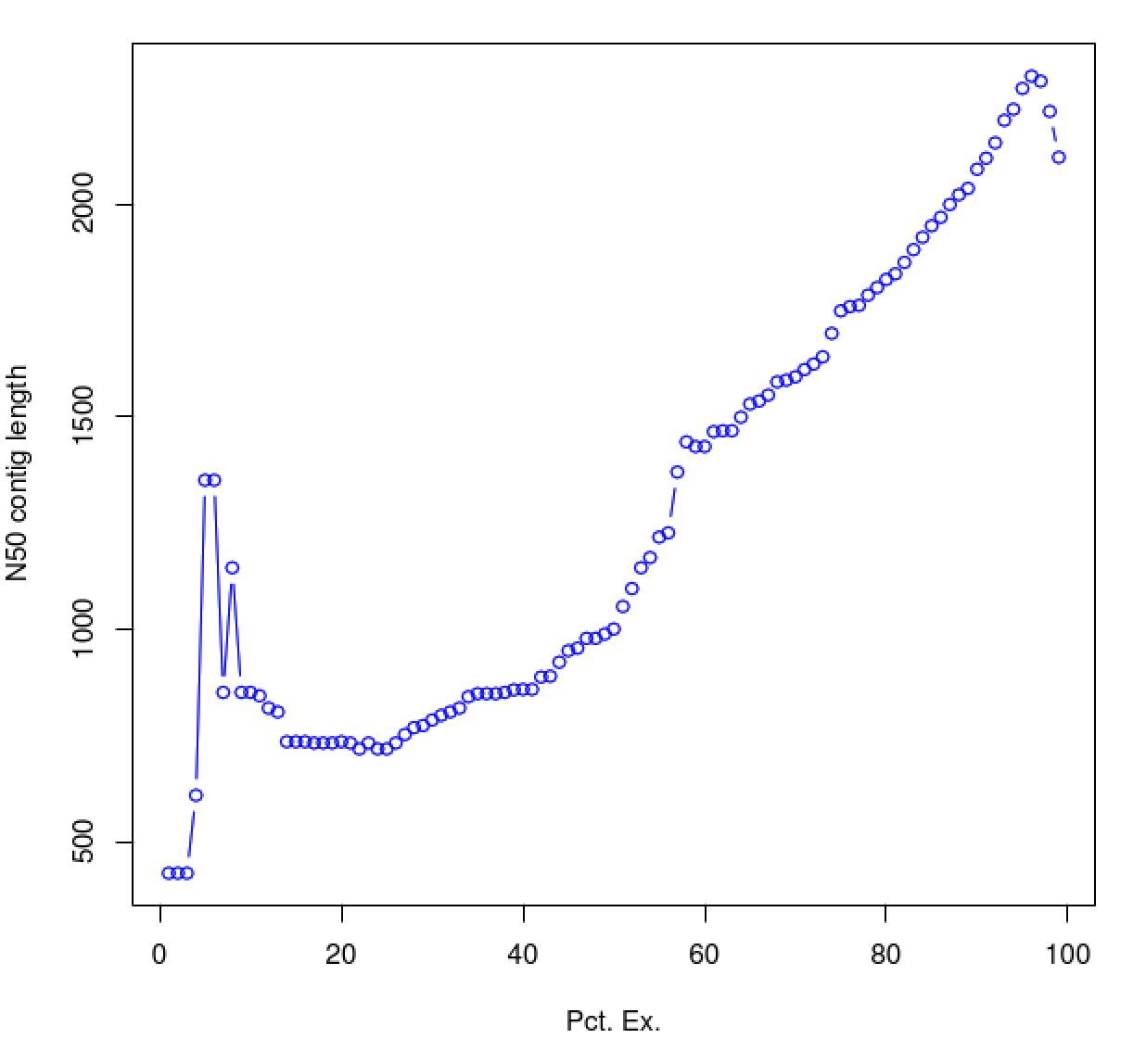
You can see that as you sequence deeper, you'll end up with an assembly that has an ExN50 peak that approaches the use of ~90% of the expression data.
Every assembled transcript is only as valid as the reads that support it. If you ever want to examine the read support for one of your favorite transcripts, you could do this using the IGV browser. Earlier, when running RSEM to estimate transcript abundance, we generated bam files containing the reads aligned to the assembled transcripts. Launch IGV on these data like so:
% igv.sh -g trinity_out_dir/Trinity.fasta \
GSNO_SRR1582648.RSEM/GSNO_SRR1582648.bowtie.csorted.bam,GSNO_SRR1582646.RSEM/GSNO_SRR1582646.bowtie.csorted.bam,GSNO_SRR1582647.RSEM/GSNO_SRR1582647.bowtie.csorted.bam,wt_SRR1582651.RSEM/wt_SRR1582651.bowtie.csorted.bam,wt_SRR1582649.RSEM/wt_SRR1582649.bowtie.csorted.bam,wt_SRR1582650.RSEM/wt_SRR1582650.bowtie.csorted.bam
Note, you could also launch IGV via clicking the icon on the desktop and then manually loading in the various input files via the menu, but that does take some time. Launching it from the command line is rather straightforward and fast.
Take some time to familiarize yourself with IGV. Look at a few transcripts and consider the read support. View the reads as pairs to examine the paired-read linkages.
A plethora of tools are currently available for identifying differentially expressed transcripts based on RNA-Seq data, and of these, edgeR is one of the most popular and most accurate. The edgeR software is part of the R Bioconductor package, and we provide support for using it in the Trinity package.
Having biological replicates for each of your samples is crucial for accurate detection of differentially expressed transcripts. In our data set, we have three biological replicates for each of our conditions, and in general, having three or more replicates for each experimental condition is highly recommended.
Create a samples.txt file containing the contents below (tab-delimited), indicating the name of the condition followed by the name of the biological replicate. Verify the contents of the file using 'cat':
Use your favorite unix text editor (eg. emacs, vim, pico, or nano) to create the file 'samples.txt' containing the tab-delimited contents below. Then verify the contents like so:
% cat samples.txt
.
GSNO GSNO_SRR1582648
GSNO GSNO_SRR1582647
GSNO GSNO_SRR1582646
WT wt_SRR1582651
WT wt_SRR1582649
WT wt_SRR1582650
Here's a trick to verify that you really have tab characters as delimiters:
% cat -te samples.txt
.
GSNO^IGSNO_SRR1582647$
GSNO^IGSNO_SRR1582646$
GSNO^IGSNO_SRR1582648$
WT^Iwt_SRR1582649$
WT^Iwt_SRR1582650$
WT^Iwt_SRR1582651$
There, you should find '^I' at the position of a tab character, and you'll find '$' at the position of a return character. This is invaluable for verifying contents of text files when the formatting has to be very precise. If your view doesn't look exactly as above, then you'll need to re-edit your file and be sure to replace any space characters you see with single tab characters, then re-examine it using the 'cat -te' approach.
The condition name (left column) can be named more or less arbitrarily but should reflect your experimental condition. Importantly, the replicate names (right column) need to match up exactly with the column headers of your RNA-Seq counts matrix.
To detect differentially expressed transcripts, run the Bioconductor package edgeR using our counts matrix:
% $TRINITY_HOME/Analysis/DifferentialExpression/run_DE_analysis.pl \
--matrix Trinity_trans.counts.matrix \
--samples_file samples.txt \
--method edgeR \
--output edgeR_trans
Examine the contents of the edgeR_trans/ directory.
% ls -ltr edgeR_trans/
.
-rw-rw-r-- 1 genomics genomics 1051 Jan 9 16:48 Trinity_trans.counts.matrix.GSNO_vs_WT.GSNO.vs.WT.EdgeR.Rscript
-rw-rw-r-- 1 genomics genomics 60902 Jan 9 16:48 Trinity_trans.counts.matrix.GSNO_vs_WT.edgeR.DE_results
-rw-rw-r-- 1 genomics genomics 18537 Jan 9 16:48 Trinity_trans.counts.matrix.GSNO_vs_WT.edgeR.DE_results.MA_n_Volcano.pdf
The files '*.DE_results' contain the output from running EdgeR to identify differentially expressed transcripts in each of the pairwise sample comparisons. Examine the format of one of the files, such as the results from comparing Sp_log to Sp_plat:
% head edgeR_trans/Trinity_trans.counts.matrix.GSNO_vs_WT.edgeR.DE_results | column -t
.
logFC logCPM PValue FDR
TRINITY_DN530_c0_g1_i1 8.98959066402695 13.1228060448362 8.32165055033775e-54 5.54221926652494e-51
TRINITY_DN589_c0_g1_i1 4.89839016049723 13.2154341051504 7.57411107973887e-53 2.52217898955304e-50
TRINITY_DN44_c0_g1_i1 2.69777851490106 14.1332252828559 5.23937214153746e-52 1.16314061542132e-49
TRINITY_DN219_c0_g1_i1 5.96230500956404 13.4919132973162 5.11512415417842e-51 8.51668171670708e-49
TRINITY_DN513_c0_g1_i1 5.67480055313841 12.8660937412604 1.54064866426519e-44 2.05214402080123e-42
TRINITY_DN494_c0_g1_i1 8.97722926993194 13.108274725098 3.6100707178792e-44 4.00717849684591e-42
TRINITY_DN365_c0_g1_i1 2.71537635410452 14.0482419858984 8.00431159168039e-41 7.61553074294163e-39
TRINITY_DN415_c0_g1_i1 6.96733684710045 12.875060733337 3.67004658844109e-36 3.05531378487721e-34
TRINITY_DN59_c0_g1_i1 -3.57509574692798 13.1852604213653 3.74452542871713e-30 2.77094881725068e-28
These data include the log fold change (logFC), log counts per million (logCPM), P- value from an exact test, and false discovery rate (FDR).
The EdgeR analysis above generated both MA and Volcano plots based on these data. Examine any of these like so:
% xpdf edgeR_trans/Trinity_trans.counts.matrix.GSNO_vs_WT.edgeR.DE_results.MA_n_Volcano.pdf
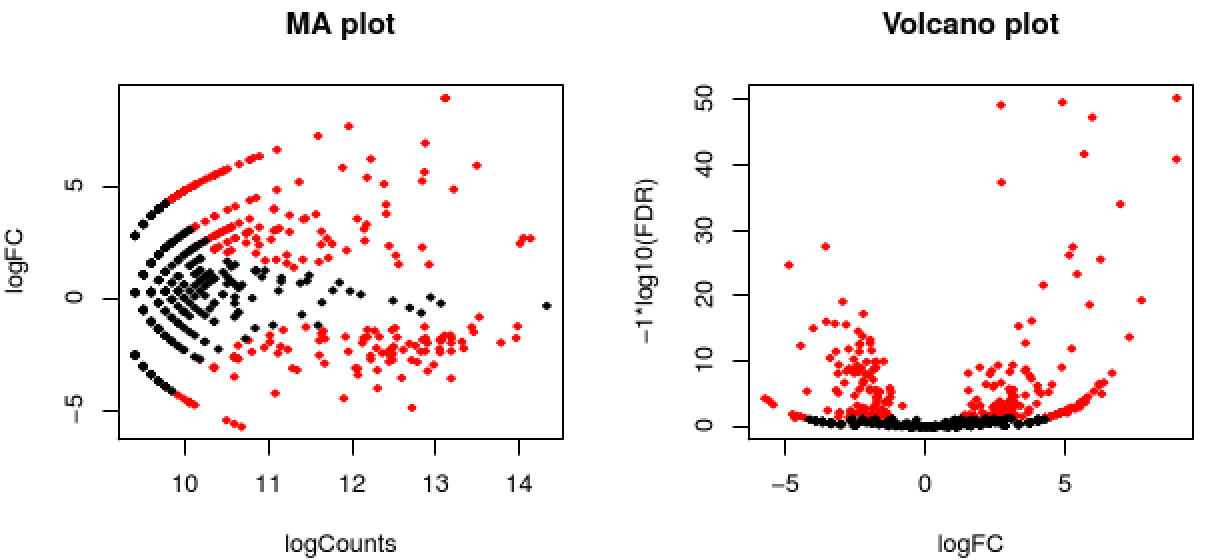
The red data points correspond to all those features that were identified as being significant with an FDR <= 0.05.
Exit the chart viewer to continue.
Trinity facilitates analysis of these data, including scripts for extracting transcripts that are above some statistical significance (FDR threshold) and fold-change in expression, and generating figures such as heatmaps and other useful plots, as described below.
Now let's perform the following operations from within the edgeR_trans/ directory. Enter the edgeR_trans/ dir like so:
% cd edgeR_trans/
Extract those differentially expressed (DE) transcripts that are at least 4-fold differentially expressed at a significance of <= 0.001 in any of the pairwise sample comparisons:
% $TRINITY_HOME/Analysis/DifferentialExpression/analyze_diff_expr.pl \
--matrix ../Trinity_trans.TMM.EXPR.matrix \
--samples ../samples.txt \
-P 1e-3 -C 2
The above generates several output files with a prefix diffExpr.P1e-3_C2', indicating the parameters chosen for filtering, where P (FDR actually) is set to 0.001, and fold change (C) is set to 2^(2) or 4-fold. (These are default parameters for the above script. See script usage before applying to your data).
Included among these files are: ‘diffExpr.P1e-3_C2.matrix’ : the subset of the FPKM matrix corresponding to the DE transcripts identified at this threshold. The number of DE transcripts identified at the specified thresholds can be obtained by examining the number of lines in this file.
% wc -l diffExpr.P1e-3_C2.matrix
.
116 diffExpr.P1e-3_C2.matrix
Note, the number of lines in this file includes the top line with column names, so there are actually 115 DE transcripts at this 4-fold and 1e-3 FDR threshold cutoff.
Also included among these files is a heatmap 'diffExpr.P1e-3_C2.matrix.log2.centered.genes_vs_samples_heatmap.pdf' as shown below, with transcripts clustered along the vertical axis and samples clustered along the horizontal axis.
% xpdf diffExpr.P1e-3_C2.matrix.log2.centered.genes_vs_samples_heatmap.pdf
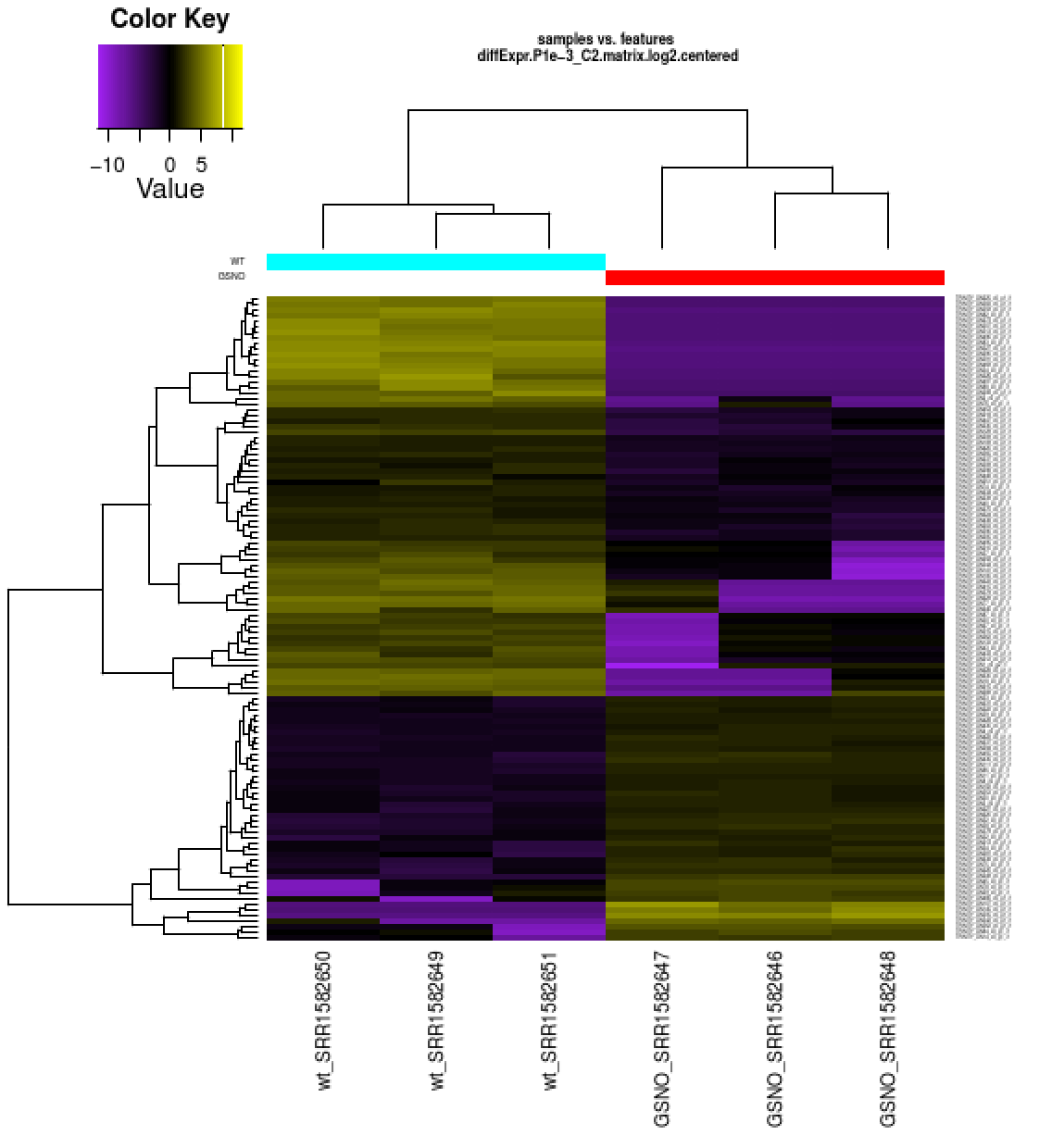
The expression values are plotted in log2 space and mean-centered (mean expression value for each feature is subtracted from each of its expression values in that row), and shows upregulated expression as yellow and downregulated expression as purple.
Exit the PDF viewer to continue.
Extract clusters of transcripts with similar expression profiles by cutting the transcript cluster dendrogram at a given percent of its height (ex. 60%), like so:
% $TRINITY_HOME/Analysis/DifferentialExpression/define_clusters_by_cutting_tree.pl \
--Ptree 60 -R diffExpr.P1e-3_C2.matrix.RData
This creates a directory containing the individual transcript clusters, including a pdf file that summarizes expression values for each cluster according to individual charts:
% xpdf diffExpr.P1e-3_C2.matrix.RData.clusters_fixed_P_60/my_cluster_plots.pdf
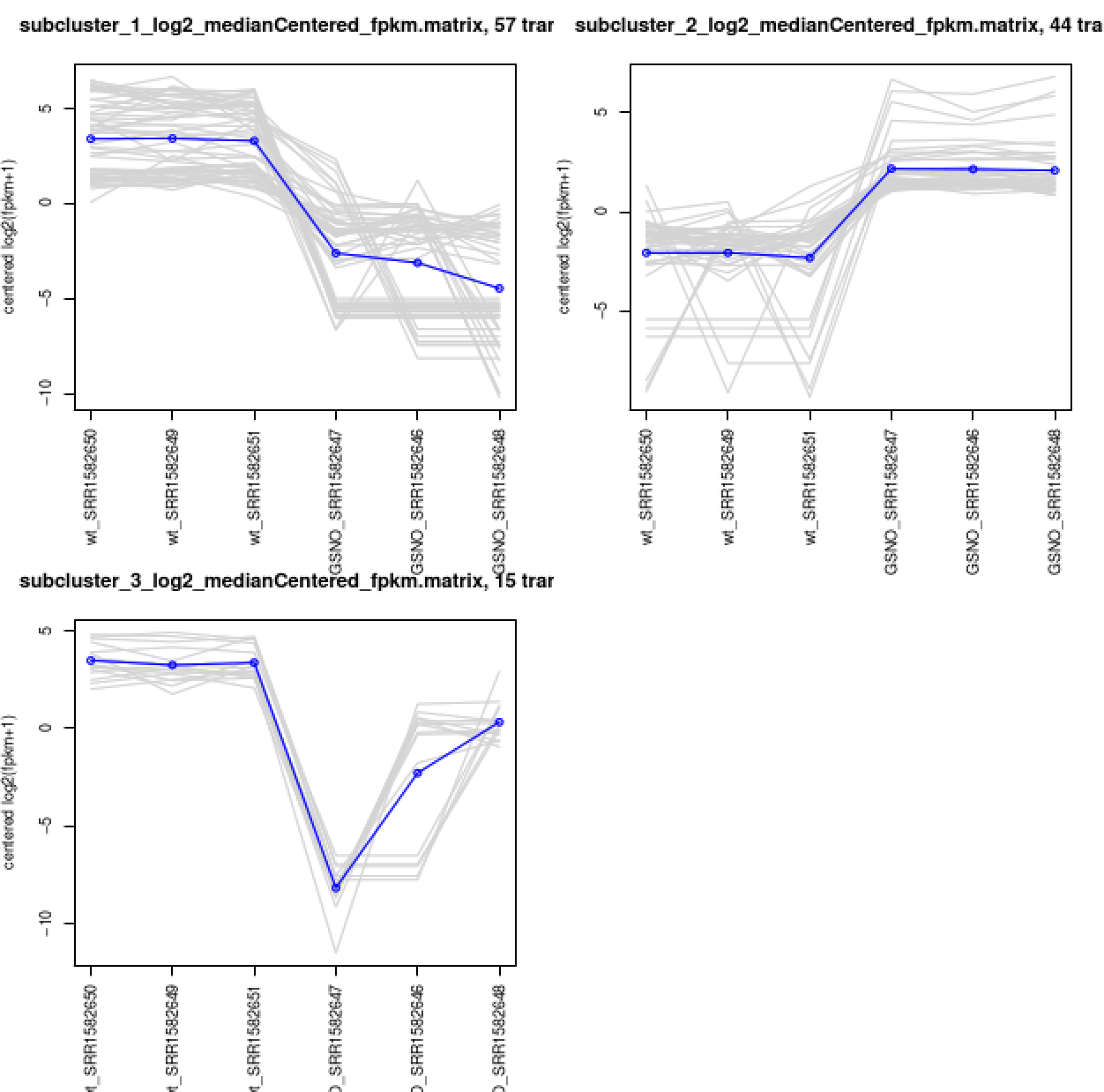
You can do all the same analyses as you did above at the gene level. For now, let's just rerun the DE detection step, since we'll need the results later on for use with TrinotateWeb. Also, it doesn't help us to study the 'gene' level data with this tiny data set (yet another disclaimer) given that all our transcripts = genes, since we didn't find any alternative splicing variants. With typical data sets, you will have alterantively spliced isoforms identified, and performing DE analysis at the gene level should provide more power for detection than at the isoform level. For more info about this, I encourage you to read this paper.
Before running the gene-level DE analysis, be sure to back out of the current edgeR_trans/ directory like so:
% cd ../
Be sure you're in your base working directory:
% pwd
.
/home/genomics/workshop_data/transcriptomics/KrumlovTrinityWorkshopJan2016
Now, run the DE analysis at the gene level like so:
% $TRINITY_HOME/Analysis/DifferentialExpression/run_DE_analysis.pl \
--matrix Trinity_genes.counts.matrix \
--samples_file samples.txt \
--method edgeR \
--output edgeR_gene
You'll now notice that the edgeR_gene/ directory exists and is populated with similar files.
% ls -ltr edgeR_gene/
Let's move on and make use of those outputs later. With your own data, however, you would normally run the same set of operations as you did above for the transcript-level DE analyses.
Now we have a bunch of transcript sequences and have identified some subset of them that appear to be biologically interesting in that they're differentially expressed between our two conditions - but we don't really know what they are or what biological functions they might represent. We can explore their potential functions by functionally annotating them using our Trinotate software and analysis protocol. To learn more about Trinotate, you can visit the Trinotate website.
Again, let's make sure that we're back in our primary working directory called 'KrumlovTrinityWorkshopJan2016':
% pwd
.
/home/genomics/workshop_data/transcriptomics/KrumlovTrinityWorkshopJan2016
If you're not in the above directory, then relocate yourself to it.
Now, create a Trinotate/ directory and relocate to it. We'll use this as our Trinotate computation workspace.
% mkdir Trinotate
% cd Trinotate
Below, we're going to run a number of different tools to capture information about our transcript sequences.
TransDecoder is a tool we built to identify likely coding regions within transcript sequences. It identifies long open reading frames (ORFs) within transcripts and scores them according to their sequence composition. Those ORFs that encode sequences with compositional properties (codon frequencies) consistent with coding transcripts are reported.
Running TransDecoder is a two-step process. First run the TransDecoder step that identifies all long ORFs.
% $TRANSDECODER_HOME/TransDecoder.LongOrfs -t ../trinity_out_dir/Trinity.fasta
Now, run the step that predicts which ORFs are likely to be coding.
% $TRANSDECODER_HOME/TransDecoder.Predict -t ../trinity_out_dir/Trinity.fasta
You'll now find a number of output files containing 'transdecoder' in their name:
% ls -1 |grep transdecoder
.
Trinity.fasta.transdecoder.bed
Trinity.fasta.transdecoder.cds
Trinity.fasta.transdecoder.gff3
Trinity.fasta.transdecoder.mRNA
Trinity.fasta.transdecoder.pep
Trinity.fasta.transdecoder_dir/
The file we care about the most here is the 'Trinity.fasta.transdecoder.pep' file, which contains the protein sequences corresponding to the predicted coding regions within the transcripts.
Go ahead and take a look at this file:
% less Trinity.fasta.transdecoder.pep
.
>TRINITY_DN102_c0_g1_i1|m.9 TRINITY_DN102_c0_g1_i1|g.9 ORF TRINITY_DN102_c0_g1_i1|g.9 \
TRINITY_DN102_c0_g1_i1|m.9 type:complete len:185 (+) TRINITY_DN102_c0_g1_i1:23-577(+)
MARYGATSTNPAKSASARGSYLRVSFKNTRETAQAINGWELTKAQKYLDQVLEHQRAIPF
RRYNSSIGRTAQGKEFGVTKARWPAKSVKFIQGLLQNAAANAEAKGLDATKLYVSHIQVN
HAPKQRRRTYRAHGRINKYESSPSHIELVVTEKEEAVAKAAEKKLVRLSSRQRGRIASQK
RITA*
>TRINITY_DN106_c0_g1_i1|m.3 TRINITY_DN106_c0_g1_i1|g.3 ORF TRINITY_DN106_c0_g1_i1|g.3 \
TRINITY_DN106_c0_g1_i1|m.3 type:5prime_partial len:149 (-) TRINITY_DN106_c0_g1_i1:38-484(-)
TNDTNESNTRTMSGNGAQGTKFRISLGLPTGAIMNCADNSGARNLYIMAVKGSGSRLNRL
PAASLGDMVMATVKKGKPELRKKVMPAIVVRQSKAWRRKDGVYLYFEDNAGVIANPKGEM
KGSAITGPVGKECADLWPRVASNSGVVV*
>TRINITY_DN109_c0_g1_i1|m.14 TRINITY_DN109_c0_g1_i1|g.14 ORF TRINITY_DN109_c0_g1_i1|g.14 \
TRINITY_DN109_c0_g1_i1|m.14 type:internal len:102 (+) TRINITY_DN109_c0_g1_i1:2-304(+)
AKVTDLRDAMFAGEHINFTEDRAVYHVALRNRANKPMKVDGVDVAPEVDAVLQHMKEFSE
QVRSGEWKGYTGKKITDVVNIGIGGSDLGPVMVTEALKHYA
>TRINITY_DN113_c0_g1_i1|m.16 TRINITY_DN113_c0_g1_i1|g.16 ORF TRINITY_DN113_c0_g1_i1|g.16 \
TRINITY_DN113_c0_g1_i1|m.16 type:3prime_partial len:123 (-) TRINITY_DN113_c0_g1_i1:2-367(-)
MSDSVTIRTRKVITNPLLARKQFVVDVLHPNRANVSKDELREKLAEAYKAEKDAVSVFGF
RTQFGGGKSTGFGLVYNSVADAKKFEPTYRLVRYGLAEKVEKASRQQRKQKKNRDKKIFG
TG
Type 'q' to exit the 'less' viewer.
There are a few items to take notice of in the above peptide file. The header lines includes the protein identifier composed of the original transcripts along with '|m.(number)'. The 'type' attribute indicates whether the protein is 'complete', containing a start and a stop codon; '5prime_partial', meaning it's missing a start codon and presumably part of the N-terminus; '3prime_partial', meaning it's missing the stop codon and presumably part of the C-terminus; or 'internal', meaning it's both 5prime-partial and 3prime-partial. You'll also see an indicator (+) or (-) to indicate which strand the coding region is found on, along with the coordinates of the ORF in that transcript sequence.
This .pep file will be used for various sequence homology and other bioinformatics analyses below.
Earlier, we ran blastx against our mini SWISSPROT datbase to identify likely full-length transcripts. Let's run blastx again to capture likely homolog information, and we'll lower our E-value threshold to 1e-5 to be less stringent than earlier.
% blastx -db ../data/mini_sprot.pep \
-query ../trinity_out_dir/Trinity.fasta -num_threads 2 \
-max_target_seqs 1 -outfmt 6 -evalue 1e-5 \
> swissprot.blastx.outfmt6
Now, let's look for sequence homologies by just searching our predicted protein sequences rather than using the entire transcript as a target:
% blastp -query Trinity.fasta.transdecoder.pep \
-db ../data/mini_sprot.pep -num_threads 2 \
-max_target_seqs 1 -outfmt 6 -evalue 1e-5 \
> swissprot.blastp.outfmt6
Using our predicted protein sequences, let's also run a HMMER search against the Pfam database, and identify conserved domains that might be indicative or suggestive of function:
% hmmscan --cpu 2 --domtblout TrinotatePFAM.out \
../../trinnotate_databases/Pfam-A.hmm \
Trinity.fasta.transdecoder.pep
Note, hmmscan might take a few minutes to run.
The signalP and tmhmm software tools are very useful for predicting signal peptides (secretion signals) and transmembrane domains, respectively.
To predict signal peptides, run signalP like so:
% signalp -f short -n signalp.out Trinity.fasta.transdecoder.pep
Take a look at the output file:
% less signalp.out
.
##gff-version 2
##sequence-name source feature start end score N/A ?
## -----------------------------------------------------------
TRINITY_DN19_c0_g1_i1|m.141 SignalP-4.0 SIGNAL 1 18 0.553 . . YES
TRINITY_DN33_c0_g1_i1|m.174 SignalP-4.0 SIGNAL 1 19 0.631 . . YES
....
How many of your proteins are predicted to encode signal peptides?
To predict transmembrane domain predictions, run tmhmm like so:
% tmhmm --short < Trinity.fasta.transdecoder.pep > tmhmm.out
Take a look at the output:
% less tmhmm.out
.
TRINITY_DN56_c0_g1_i1|m.126 len=303 ExpAA=126.72 First60=16.21 PredHel=6 Topology=o44-66i100-119o129-148i155-177o187-209i222-241o
TRINITY_DN56_c0_g2_i1|m.127 len=262 ExpAA=126.02 First60=23.86 PredHel=6 Topology=o5-24i59-78o88-107i114-136o146-168i181-200o
Those entries that have results contain output like so 'Topology=o5-24i59-78o88-107i114-136o146-168i181-200o' are the predicted transmembrane proteins. The number of helices in the transmembrane domain is listed under PredHel=(number) and the topology indicates the region within the protein sequence that spans the membrane from (i) inside to (o) outside the cell.
Generating a Trinotate annotation report involves first loading all of our bioinformatics computational results into a Trinotate SQLite database. The Trinotate software provides a boilerplate SQLite database called 'Trinotate.sqlite' that comes pre-populated with a lot of generic data about SWISSPROT records and Pfam domains (and is a pretty large file consuming several GB). Below, we'll populate this database with all of our bioinformatics computes and our expression data. You'll find your 'Trinotate.sqlite' database in your base working directory for this workshop (ie. '/..../KrumlovTrinityWorkshopJan2016/Trinotate.sqlite').
As a sanity check, be sure you're currently located in your 'Trinotate/' working directory.
% pwd
.
/home/genomics/workshop_data/transcriptomics/KrumlovTrinityWorkshopJan2016/Trinotate
Load your Trinotate.sqlite database with your Trinity transcripts and predicted protein sequences:
% $TRINOTATE_HOME/Trinotate ../Trinotate.sqlite init \
--gene_trans_map ../trinity_out_dir/Trinity.fasta.gene_trans_map \
--transcript_fasta ../trinity_out_dir/Trinity.fasta \
--transdecoder_pep Trinity.fasta.transdecoder.pep
Load in the various outputs generated earlier:
% $TRINOTATE_HOME/Trinotate ../Trinotate.sqlite \
LOAD_swissprot_blastx swissprot.blastx.outfmt6
% $TRINOTATE_HOME/Trinotate ../Trinotate.sqlite \
LOAD_swissprot_blastp swissprot.blastp.outfmt6
% $TRINOTATE_HOME/Trinotate ../Trinotate.sqlite LOAD_pfam TrinotatePFAM.out
% $TRINOTATE_HOME/Trinotate ../Trinotate.sqlite LOAD_tmhmm tmhmm.out
% $TRINOTATE_HOME/Trinotate ../Trinotate.sqlite LOAD_signalp signalp.out
% $TRINOTATE_HOME/Trinotate ../Trinotate.sqlite report > Trinotate.xls
View the report
% less Trinotate.xls
.
#gene_id transcript_id sprot_Top_BLASTX_hit TrEMBL_Top_BLASTX_hit RNAMMER prot_id prot_coords sprot_Top_BLASTP_hit TrEMBL_Top_BLASTP_hit Pfam SignalP TmHMM eggnog gene_ontology_blast gene_ontology_pfam transcript peptide
TRINITY_DN144_c0_g1 TRINITY_DN144_c0_g1_i1 PUT4_YEAST^PUT4_YEAST^Q:1-198,H:425-490^74.24%ID^E:4e-29^RecName: Full=Proline-specific permease;^Eukaryota; Fungi; Dikarya; Ascomycota; Saccharomycotina; Saccharomycetes; Saccharomycetales; Saccharomycetaceae; Saccharomyces . . . .
. . . . . COG0833^permease GO:0016021^cellular_component^integral component of membrane`GO:0005886^cellular_component^plasma membrane`GO:0015193^molecular_function^L-proline transmembrane transporter activity`GO:0015175^molecular_function^neutral amino acid transmembrane transporter activity`GO:0015812^biological_process^gamma-aminobutyric acid transport`GO:0015804^biological_process^neutral amino acid transport`GO:0035524^biological_process^proline transmembrane transport`GO:0015824^biological_process^proline transport . . .
TRINITY_DN179_c0_g1 TRINITY_DN179_c0_g1_i1 ASNS1_YEAST^ASNS1_YEAST^Q:1-168,H:158-213^82.14%ID^E:5e-30^RecName: Full=Asparagine synthetase [glutamine-hydrolyzing] 1;^Eukaryota; Fungi; Dikarya; Ascomycota; Saccharomycotina; Saccharomycetes; Saccharomycetales; Saccharomycetaceae; Saccharomyces .
. . . . . . . . COG0367^asparagine synthetase GO:0004066^molecular_function^asparagine synthase (glutamine-hydrolyzing) activity`GO:0005524^molecular_function^ATP binding`GO:0006529^biological_process^asparagine biosynthetic process`GO:0006541^biological_process^glutamine metabolic process`GO:0070981^biological_process^L-asparagine biosynthetic process . . .
TRINITY_DN159_c0_g1 TRINITY_DN159_c0_g1_i1 ENO2_CANGA^ENO2_CANGA^Q:2-523,H:128-301^100%ID^E:4e-126^RecName: Full=Enolase 2;^Eukaryota; Fungi; Dikarya; Ascomycota; Saccharomycotina; Saccharomycetes; Saccharomycetales; Saccharomycetaceae; Nakaseomyces; Nakaseomyces/Candida clade . . TRINITY_DN159_c0_g1_i1|m.1 2-523[+] ENO2_CANGA^ENO2_CANGA^Q:1-174,H:128-301^100%ID^E:3e-126^RecName: Full=Enolase 2;^Eukaryota; Fungi; Dikarya; Ascomycota; Saccharomycotina; Saccharomycetes; Saccharomycetales; Saccharomycetaceae; Nakaseomyces; Nakaseomyces/Candida clade . PF00113.17^Enolase_C^Enolase, C-terminal TIM barrel domain^18-174^E:9.2e-79 . . . GO:0005829^cellular_component^cytosol`GO:0000015^cellular_component^phosphopyruvate hydratase complex`GO:0000287^molecular_function^magnesium ion binding`GO:0004634^molecular_function^phosphopyruvate hydratase activity`GO:0006096^biological_process^glycolytic process GO:0000287^molecular_function^magnesium ion binding`GO:0004634^molecular_function^phosphopyruvate hydratase activity`GO:0006096^biological_process^glycolytic process`GO:0000015^cellular_component^phosphopyruvate hydratase complex . .
...
The above file can be very large. It's often useful to load it into a spreadsheet software tools such as MS-Excel. If you have a transcript identifier of interest, you can always just 'grep' to pull out the annotation for that transcript from this report. We'll use TrinotateWeb to interactively explore these data in a web browser below.
Let's use the annotation attributes for the transcripts here as 'names' for the transcripts in the Trinotate database. This will be useful later when using the TrinotateWeb framework.
% $TRINOTATE_HOME/util/annotation_importer/import_transcript_names.pl \
../Trinotate.sqlite Trinotate.xls
Nothing exciting to see in running the above command, but know that it's helpful for later on.
Notice that the report above contains Gene Ontology identifiers mapped to transcripts. These are derived from SWISSPROT records of matching proteins as well as assigned according to Pfam domain content. The Gene Ontology structured classifications will allow us to explore whether we have statistically significant functional enrichment based on gene sets - such as for those genes that were identified to be differentially expressed.
Generate a listing of all GO identifiers assigned to each transcript like so:
% $TRINOTATE_HOME/util/extract_GO_assignments_from_Trinotate_xls.pl \
--Trinotate_xls Trinotate.xls -T -I > Trinotate.xls.gene_ontology
If you see warning messages 'cannot parse parent info', simply ignore them.
and take a look at the output file:
% less Trinotate.xls.gene_ontology
.
TRINITY_DN101_c0_g1_i1 GO:0003674,GO:0003824,GO:0005488,GO:0005506,GO:0005575,GO:0005759,GO:0005829,GO:0006810,GO:0006950,GO:0006979,GO:0008150,GO:0008941,GO:0009636,GO:0009987,GO:0015669,GO:0015671,GO:0016491,GO:0016661,GO:0016662,GO:0016705,GO:0016708,GO:0016966,GO:0019825,GO:0020037,GO:0031974,GO:0033554,GO:0034599,GO:0042221,GO:0043167,GO:0043169,GO:0043233,GO:0044422,GO:0044424,GO:0044429,GO:0044444,GO:0044446,GO:0044464,GO:0044699,GO:0044765,GO:0046872,GO:0046906,GO:0046914,GO:0050896,GO:0051179,GO:0051213,GO:0051234,GO:0051716,GO:0070013,GO:0070887,GO:0097159,GO:1901363,GO:1902578
TRINITY_DN102_c0_g1_i1 GO:0002181,GO:0003674,GO:0003735,GO:0005198,GO:0005575,GO:0005840,GO:0006412,GO:0008150,GO:0008152,GO:0009058,GO:0009059,GO:0009987,GO:0015934,GO:0019538,GO:0022625,GO:0030529,GO:0032991,GO:0034645,GO:0043170,GO:0043226,GO:0043228,GO:0043229,GO:0043232,GO:0044237,GO:0044238,GO:0044249,GO:0044260,GO:0044267,GO:0044391,GO:0044422,GO:0044424,GO:0044444,GO:0044445,GO:0044446,GO:0044464,GO:0071704,GO:1901576
TRINITY_DN103_c0_g1_i1 GO:0003674,GO:0003824,GO:0004130,GO:0004601,GO:0005488,GO:0005575,GO:0005759,GO:0005829,GO:0006950,GO:0006979,GO:0008150,GO:0016209,GO:0016491,GO:0016684,GO:0020037,GO:0031974,GO:0043167,GO:0043169,GO:0043233,GO:0044422,GO:0044424,GO:0044429,GO:0044444,GO:0044446,GO:0044464,GO:0046872,GO:0046906,GO:0050896,GO:0070013,GO:0097159,GO:1901363
...
Now that we have our list of transcript-to-GO assignment mappings, let's go back to the edgeR directory and explore functional enrichments of our DE transcripts:
% cd ../edgeR_trans
We'll use the GOseq software to test for GO enrichment. One of the key merits of GOseq is that it takes into account biases in transcript feature lengths according to functional categories (some functional categories have many long or short proteins, which biases their representation as detected in DE data). Generate a file containing the lengths of each of our assembled transcripts like so:
% $TRINITY_HOME/util/misc/fasta_seq_length.pl \
../trinity_out_dir/Trinity.fasta > Trinity.seqLengths
Take a glance at the file we just generated:
% less Trinity.seqLengths
.
fasta_entry length
TRINITY_DN144_c0_g1_i1 199
TRINITY_DN179_c0_g1_i1 169
TRINITY_DN159_c0_g1_i1 524
TRINITY_DN153_c0_g1_i1 182
TRINITY_DN130_c0_g1_i1 254
Now, run GOseq on our subset of transcripts that were identified as significantly DE given our earlier chosen thresholds. We'll start with the subset of transcripts that were identified as significantly upregulated under the GSNO treatment:
% $TRINITY_HOME/Analysis/DifferentialExpression/run_GOseq.pl \
--genes_single_factor Trinity_trans.counts.matrix.GSNO_vs_WT.edgeR.DE_results.P1e-3_C2.GSNO-UP.subset \
--GO_assignments ../Trinotate/Trinotate.xls.gene_ontology \
--lengths Trinity.seqLengths
This will generate two output files, one containing GO categories enriched among that set of DE transcripts (.GSNO-UP.subset.GOseq.enriched), and another containing GO categores that are depleted (.GSNO-UP.subset.GOseq.depleted).
Examine the contents of these files using 'less'.
Note, the small number of reads we are using for the assembly and DE analysis is going to hinder our sensitivity for detection of DE transcripts and this will funnel into a lack of sensitivity in picking up functional category enrichments. We do identify functional category enrichments here, but the most informative findings would require assembling the original millions of RNA-Seq reads to find them.
You can run the same GOseq functional enrichment analysis on those transcripts that were found significantly upregulated under the rich media growth conditions.
% $TRINITY_HOME/Analysis/DifferentialExpression/run_GOseq.pl \
--genes_single_factor Trinity_trans.counts.matrix.GSNO_vs_WT.edgeR.DE_results.P1e-3_C2.WT-UP.subset \
--GO_assignments ../Trinotate/Trinotate.xls.gene_ontology \
--lengths Trinity.seqLengths
See which files were just generated as a result:
% ls -ltr
and view each to see if there's anything interesting. Remember the caveat mentioned above about not being well powered in having used our small example set of reads.
Earlier, we generated large sets of tab-delimited files containg lots of data - annotations for transcripts, matrices of expression values, lists of differentially expressed transcripts, etc. We also generated a number of plots in PDF format. These are all useful, but they're not interactive and it's often difficult and cumbersome to extract information of interest during a study. We're developing TrinotateWeb as a web-based interactive system to solve some of these challenges. TrinotateWeb provides heatmaps and various plots of expression data, and includes search functions to quickly access information of interest. Below, we will populate some of the additional information that we need into our Trinotate database, and then run TrinotateWeb and start exploring our data in a web browser.
Let's change our working directory back to the Trinotate/ workspace:
% cd ../Trinotate
Now, load in the transcript expression data stored in the matrices we built earlier:
% $TRINOTATE_HOME/util/transcript_expression/import_expression_and_DE_results.pl \
--sqlite ../Trinotate.sqlite \
--transcript_mode \
--samples_file ../samples.txt \
--count_matrix ../Trinity_trans.counts.matrix \
--fpkm_matrix ../Trinity_trans.TMM.EXPR.matrix
Import the DE results from our edgeR_trans/ directory:
% $TRINOTATE_HOME/util/transcript_expression/import_expression_and_DE_results.pl \
--sqlite ../Trinotate.sqlite \
--transcript_mode \
--samples_file ../samples.txt \
--DE_dir ../edgeR_trans
and Import the clusters of transcripts we extracted earlier based on having similar expression profiles across samples:
% $TRINOTATE_HOME/util/transcript_expression/import_transcript_clusters.pl \
--sqlite ../Trinotate.sqlite \
--group_name DE_all_vs_all \
--analysis_name diffExpr.P1e-3_C2.matrix.RData.clusters_fixed_P_60 \
../edgeR/diffExpr.P1e-3_C2.matrix.RData.clusters_fixed_P_60/*matrix
And now we'll do the same for our gene-level expression and DE results:
% $TRINOTATE_HOME/util/transcript_expression/import_expression_and_DE_results.pl \
--sqlite ../Trinotate.sqlite \
--component_mode \
--samples_file ../samples.txt \
--count_matrix ../Trinity_genes.counts.matrix \
--fpkm_matrix ../Trinity_genes.TMM.EXPR.matrix
% $TRINOTATE_HOME/util/transcript_expression/import_expression_and_DE_results.pl \
--sqlite ../Trinotate.sqlite \
--component_mode \
--samples_file ../samples.txt \
--DE_dir ../edgeR_gene
Note, in the above gene-loading commands, the term 'component' is used. 'Component' is just another word for 'gene' in the realm of Trinty.
At this point, the Trinotate database should be fully populated and ready to be used by TrinotateWeb.
TrinotateWeb is web-based software and runs locally on the same hardware we've been running all our computes (as opposed to your typical websites that you visit regularly, such as facebook). Launch the mini webserver that drives the TrinotateWeb software like so:
% $TRINOTATE_HOME/run_TrinotateWebserver.pl 3000
Now, visit the following URL in Google Chrome: http://localhost:3000/cgi-bin/index.cgi
You should see a web form like so:
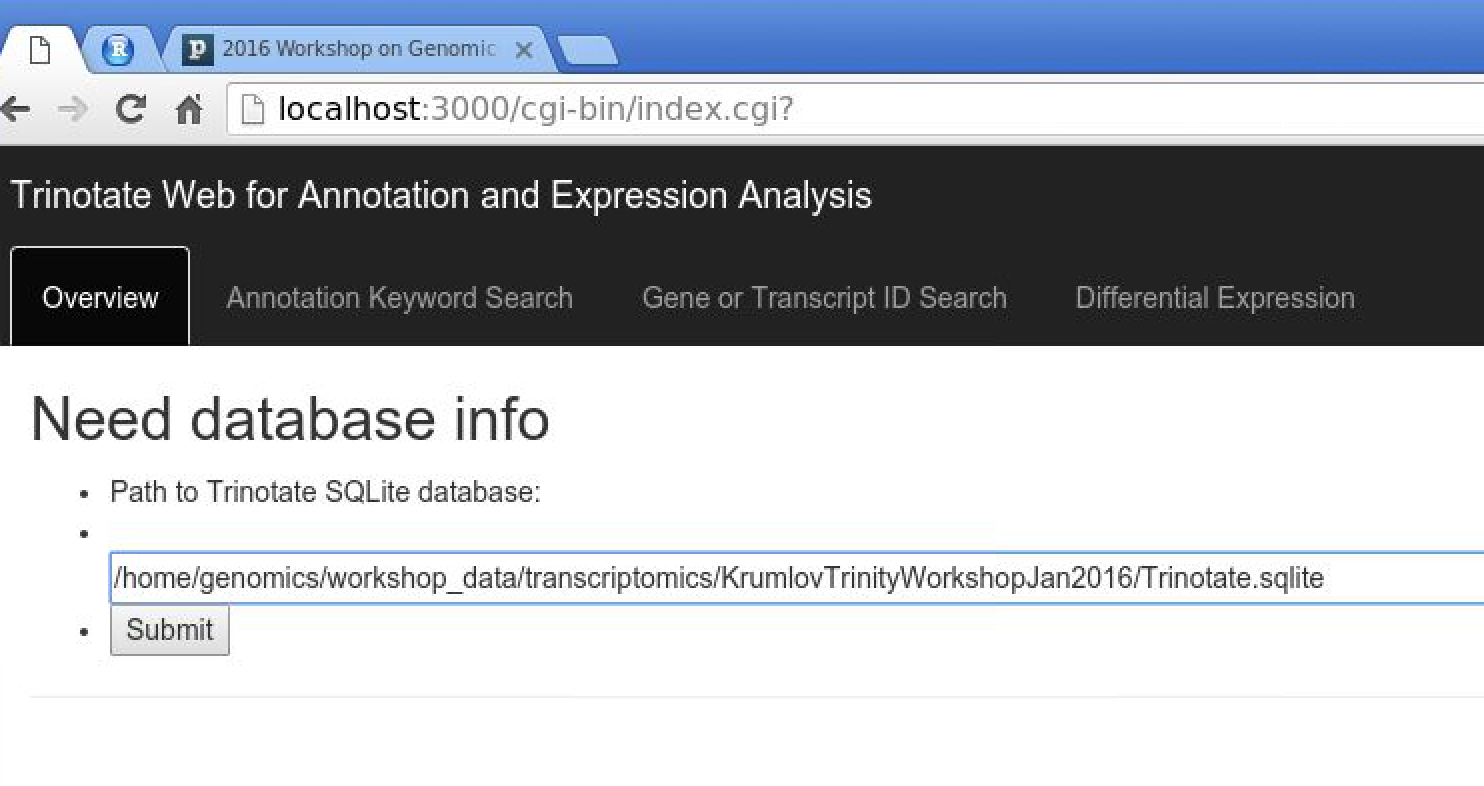
In the text box, put the path to your Trinotate.sqlite database, as shown above ("/home/genomics/workshop_data/transcriptomics/KrumlovTrinityWorkshopJan2016/Trinotate.sqlite"). Click 'Submit'.
You should now have TrinotateWeb running and serving the content in your Trinotate database:
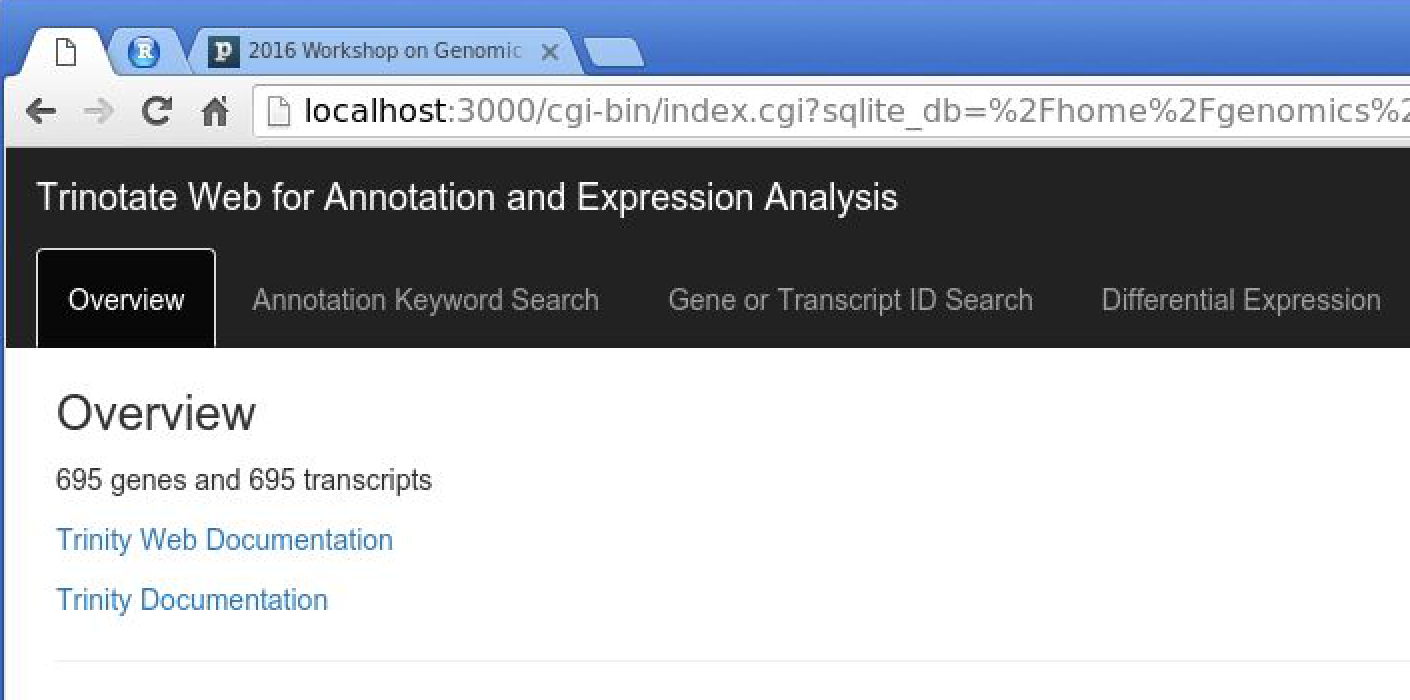
Take some time to click the various tabs and explore what's available.
eg. Under 'Annotation Keyword Search', search for 'transporter'
eg. Under 'Differential Expression', examine your earlier-defined transcript clusters. Also, launch MA or Volcano plots to explore the DE data.
We will explore TrinotateWeb functionality together as a group.
If you've gotten this far, hurray!!! Congratulations!!! You've now experienced the full tour of Trinity and TrinotateWeb. Visit our web documentation at http://trinityrnaseq.github.io, and join our Google group to become part of the ever-growing Trinity user community.