diff --git a/README.md b/README.md
new file mode 100644
index 0000000..180c9b8
--- /dev/null
+++ b/README.md
@@ -0,0 +1,138 @@
+**Mabs** is a genome assembly tool which optimizes parameters of genome assemblers Hifiasm and Flye.
+Briefly, Mabs works as follows:
+1) It makes a series of genome assemblies by Hifiasm or Flye, using different values of parameters of these programs. Mabs uses special tricks to accelerate the assembly process.
+2) For each genome assembly, Mabs evaluates the quality of BUSCO genes' assembly using a special metric that I call "AG". For how AG is calculated, see [calculate_AG](#internal_link_to_calculate_AG).
+3) The genome assembly with the largest AG is condidered the best.
+
+Mabs is, on average, 3 times slower than Hifiasm or Flye, but usually produces better or equal assemblies. For details, see a link to BioRxiv will be here in a few days.
+
+## Table of Contents
+
+- [Installation](#internal_link_to_Installation)
+- [How to use](#internal_link_to_How_to_use)
+ - [Mabs-hifiasm](#internal_link_to_Mabs-hifiasm)
+ - [Mabs-flye](#internal_link_to_Mabs-flye)
+ - [The output of Mabs](#internal_link_to_The_output_of_Mabs)
+ - [Testing Mabs-hifiasm and Mabs-flye](#internal_link_to_Testing_Mabs-hifiasm_and_Mabs-flye)
+- [calculate_AG](#internal_link_to_calculate_AG)
+- [Questions and Answers](#internal_link_to_Questions_and_Answers)
+
+
+## Installation
+Mabs requires Python 3, Perl 5, GCC, Zlib-dev, Make.
+To install Mabs, download the latest version from [Releases](https://github.com/shelkmike/Mabs/releases), then extract the archive and run
+`bash install.sh`
+
+
+## How to use
+Two main components of Mabs are Mabs-hifiasm and Mabs-flye. Mabs-hifiasm works as a parameter optimizer of Hifiasm, while Mabs-fly works as a parameter optimizer of Flye.
+
+#### a) Mabs-hifiasm
+Mabs-hifiasm is intended for PacBio HiFi (also known as CCS) reads. Also, it can be used for very accurate (accuracy ≥99%) Nanopore reads, as their characteristics are similar to characteristics of HiFi reads.
+To run Mabs-hifiasm, a user should provide two values:
+1. A path to reads, via the option "--pacbio_hifi_reads".
+2. A BUSCO dataset. In the process of parameters optimization, Mabs uses a BUSCO dataset. The dataset can be provided using either the option "--download_busco_dataset", or the option "--local_busco_dataset".
+The option "--download_busco_dataset" requires a filename from https://mikeshelk.site/Data/BUSCO_datasets/Latest/ . It is recommended to use the most taxonomically narrow dataset. For example, if you assemble a drosophila genome, use "--download_busco_dataset diptera_odb10.2020-08-05.tar.gz".
+Alternatively, you can download a dataset to your computer manually, and use the option "--local_busco_dataset". For example, "--local_busco_dataset /home/username/Work/diptera_odb10.2020-08-05.tar.gz".
+
+To see the full list of options, run
+`mabs-hifiasm.py --help`
+
+Since Mabs-hifiasm is based on Hifiasm (https://github.com/chhylp123/hifiasm), it can use paired-end Hi-C reads in addition to long reads. Provide trimmed Hi-C reads with options "--short_hi-c_reads_R1" and "--short_hi-c_reads_R2".
+
+Examples of using Mabs-hifiasm.
+
+Example 1:
+`mabs-hifiasm.py --pacbio_hifi_reads hifi_reads.fastq --download_busco_dataset eudicots_odb10.2020-09-10.tar.gz --threads 40`
+
+Example 2:
+`mabs-hifiasm.py --pacbio_hifi_reads hifi_reads.fastq --short_hi-c_reads_R1 hi-c_reads_trimmed_R1.fastq --short_hi-c_reads_R2 hi-c_reads_trimmed_R2.fastq --download_busco_dataset diptera_odb10.2020-08-05.tar.gz --threads 40`
+
+
+#### b) Mabs-flye
+Mabs-flye is intended for Nanopore reads and PacBio CLR reads (also known as "old PacBio reads"). Similarly to Mabs-hifiasm, Mabs-flye requires two values:
+1. A path to reads, provided via options "--nanopore_reads", "--pacbio_clr_reads" or "--pacbio_hifi_reads". If you have several read datatets created by different technologies, these options can be used simultaneously. Keep in mind that if you have only HiFi reads, it's better to use Mabs-hifiasm.
+2. A path to a BUSCO dataset, provided via options "--download_busco_dataset" or "--local_busco_dataset". For details, see "2." in the description of Mabs-hifiasm above.
+
+To see the full list of options, run
+`mabs-flye.py --help`
+
+Examples of using Mabs-flye.
+
+Example 1:
+`mabs-flye.py --nanopore_reads nanopore_reads.fastq --download_busco_dataset eudicots_odb10.2020-09-10.tar.gz --threads 40`
+
+Example 2:
+`mabs-flye.py --nanopore_reads nanopore_reads.fastq --pacbio_hifi_reads pacbio_hifi_reads.fastq --download_busco_dataset diptera_odb10.2020-08-05.tar.gz --threads 40`
+
+
+#### c) The output of Mabs
+Both Mabs-hifiasm and Mabs-flye have a similar output structure. Both of them create a folder which, by default, is named "Mabs_results". The name can be changed via the "--output_folder" option. The two main files that a user may need are:
+1) ./Mabs_results/mabs_logs.txt
+This file contains information on how Mabs-hifiasm or Mabs-flye run and whether they encountered any problems.
+2) ./Mabs_results/The_best_assembly/assembly.fasta
+These are the contigs you need.
+
+
+#### d) Testing Mabs-hifiasm and Mabs-flye
+If you are not sure whether Mabs-hifiasm and Mabs-flye have been installed properly, you can run
+`mabs-hifiasm.py --run_test`
+or
+`mabs-flye.py --run_test`
+
+These two commands assemble the first chromosome of Saccharomyces cerevisiae, which is approximately 200 kbp. If after the assembly finishes you see a file ./Mabs_results/The_best_assembly/assembly.fasta which is slightly larger than 200 KB, Mabs works correctly.
+
+
+## calculate_AG
+Besides Mabs-hifiasm and Mabs-flye, Mabs contains a third tool, named calculate_AG. It's purpose is to assess genome assembly quality.
+
+calculate_AG is used internally by Mabs-hifiasm and Mabs-flye, but also can be used externally if a user wants to assess the quality of some assembly.
+The main concept in calculate_AG is "AG", which is a metric of gene assembly quality used by Mabs. "AG" is short for "Accurately assembled Genes". It is calculated as a sum of the following two values:
+a) The number of genes in single-copy BUSCO orthogroups.
+b) The number of genes in true multicopy orthogroups. "True multicopy" means that there are more than one gene in these orthogroups not because of assembly errors, but because these genes are actual paralogs. In contrast, the number of genes in false multicopy orthogroups (the orthogroups where genes duplications are because of assembly errors) are not included in AG.
+AG, in my opinion, may be a better metric of gene assembly quality than BUSCO results, because BUSCO does not differentiate true multicopy orthogroups and false multicopy orthogroups, combining them into a single "D" category.
+A frequent cause of false multicopy orthogroups are haplotypic duplications, when two alleles of a gene are erroneously assembled as paralogs.
+calculate_AG differentiates true multicopy orthogroups from false multicopy orthogroups based on gene coverage, since if a duplication is an assembly error, the gene coverage should be decreased.
+
+Basically, the larger AG is, the better the assembly is.
+
+If a user wants to calculate AG for some genome assembly, he can use a command like:
+`calculate_AG.py --assembly contigs.fasta --nanopore nanopore_reads.fastq --local_busco_dataset /mnt/lustre/username/Datasets/eudicots_odb10 --threads 40`
+
+For more options, run
+`calculate_AG.py --help`
+
+The main file produced by calculate_AG is ./AG_calculation_results/AG.txt . It contains a single number which is the AG.
+In addition, calculate_AG produces figures gene_coverage_distribution.svg and gene_coverage_distribution.png which look like this:
+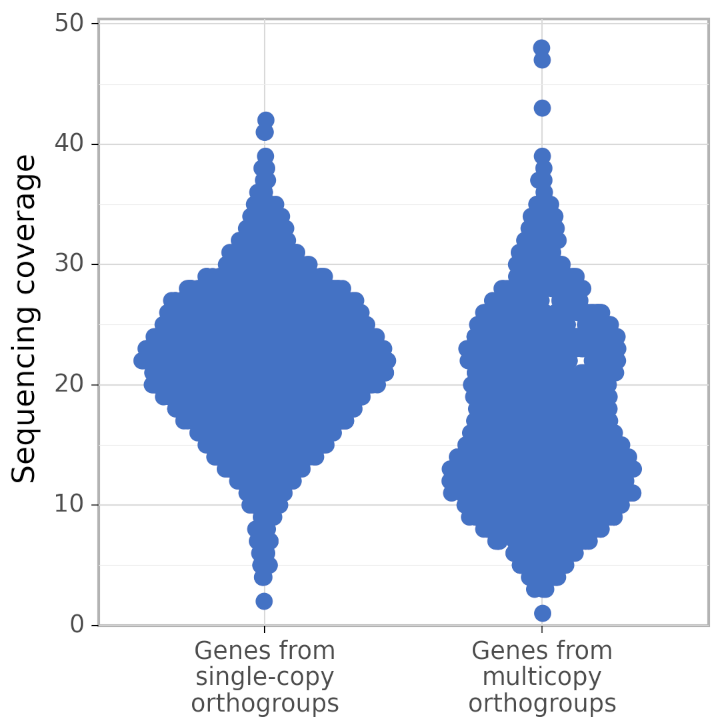
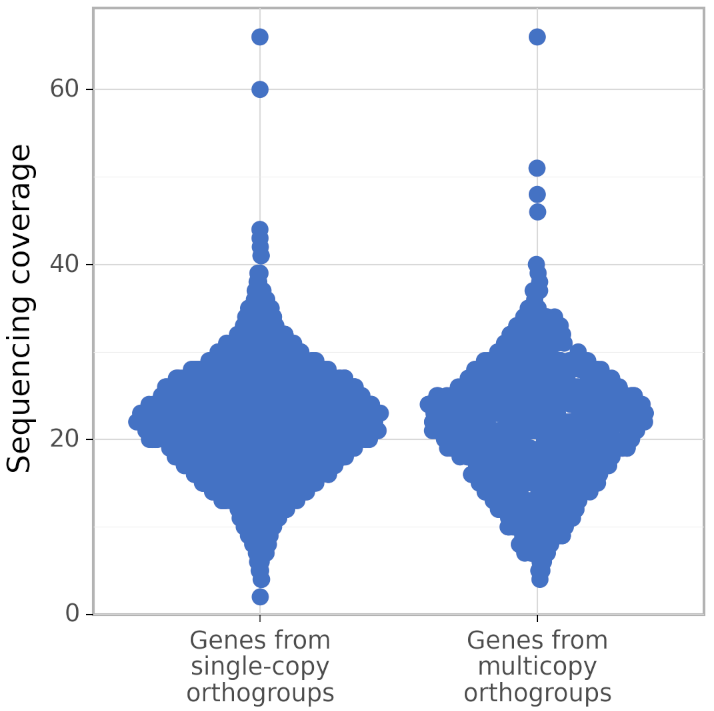
+These type of diagrams are called sinaplots, see https://cran.r-project.org/web/packages/sinaplot/vignettes/SinaPlot.html . The sinaplots produced by calculate_AG help to evaluate the assembly quality visually. While the coverage distribution of genes from single-copy orthogroups is unimodal, the coverage distrubution of genes from multicopy orthogroups can be bimodal because genes that were erroneously duplicated have twice as low coverage as they should have. In the perfect assembly, the coverage distribution of genes from multicopy orthogroups is identical to the coverage distribution of genes from single-copy orthogroups. The picture above is for a rather bad assembly. Below is the picture made by calculate_AG for a better assembly of the same genome:
+
+
+The recommended usage of calculate_AG is to compare the quality of assemblies of a single genome made by different genome assemblers, or made by a single assembler with different parameters. Besides the value of AG (in the file ./AG_calculation_results/AG.txt), calculate_AG also provide the exact numbers of genes in single-copy orthogroups, in true multicopy orthogroups, and in false multicopy orthogroups; the corresponding values can be found at the end of the file ./AG_calculation_results/logs.txt.
+
+
+## Questions and Answers
+1. Should assemblies produced by Mabs be polished afterwards?
+The assemblies made by Mabs-hifiasm are accurate already. The assemblies made by Mabs-flye require polishing by accurate reads. "Accurate reads" are reads of Illumina, MGI, or PacBio HiFi. Good programs for polishing are, for example, [HyPo](https://github.com/kensung-lab/hypo), [POLCA](https://github.com/alekseyzimin/masurca), [Racon](https://github.com/lbcb-sci/racon).
+2. How to assemble a genome using high-accuracy Nanopore reads?
+If you have Nanopore reads with really high accuracy (≥99%), I advise to try both Mabs-hifiasm and Mabs-flye.
+3. What is the program "Modified_hifiasm" used by Mabs?
+Modified_hifiasm is a special version of Hifiasm, where I added an option "--only-primary". With this option, Modified_hifiasm stops after creating the file with the primary assembly. Usage of Modified_hifiasm makes Mabs-hifiasm faster then when using the original Hifiasm.
+4. Is it worth to use Mabs if I don't expect a high number of haplotypic duplications?
+Though the primary purpose of Mabs is creation of assemblies with few haplotypic duplications, it may be useful even if you don't expect many haplotypic duplications. Since Mabs optimizes parameters of Hifiasm or Flye to maximize the gene assembly quality, in most cases it will produce assemblies better than or equal to Hifiasm or Flye.
+5. Can Mabs be used to assemble metagenomes?
+No. When evaluating which genes were assembled correctly and which were assembled incorrectly, Mabs relies on their coverage. In a metagenomic sequencing different genomes have different coverage, which makes Mabs useless.
+6. Can Mabs be used to assemble haploid genomes, for example bacterial?
+Yes. Though, I don't expect Mabs to be much better than Hifiasm and Flye for haploid genomes since haploid genome assemblies cannot have haplotypic duplications.
+7. Can Mabs-hifiasm perform a trio binning assembly as Hifiasm does?
+No. If you want me to add this functionality, let me know via [Issues](https://github.com/shelkmike/Mabs/issues).
+8. The option "--download_busco_dataset" fails to download a BUSCO dataset. What should I do?
+This can happen if http://mikeshelk.site and, consequently, http://mikeshelk.site/Data/BUSCO_datasets/Latest/ is currently not accessible for some reason. To deal with this problem, manually download a file from http://busco-data.ezlab.org/v5/data/lineages/ and provide it to Mabs via the option "--local_busco_dataset".
+9. What does "Mabs" mean?
+Funny to say, but "Mabs" means "Miniasm-based Assembler which maximizes Busco Score". That's because:
+a) Mabs 1 was based on Miniasm instead of Hifiasm and Flye.
+Miniasm takes as input a set of read overlaps produced by a program like Minimap2. Provided a file with overlaps, Miniasm performs assembly very quickly. The prominent speed of Miniasm allows to explore the parameter space more thoroughly than when using Hifiasm or Flye, which are 1-2 orders of magnitude slower. However, I later realized that the algorithm of Miniasm is inferior to the algorithms of Hifiasm and Flye, and even a more thorough exploration of a parameter space usually doesn't make Miniasm assemblies better than assemblies of Hifiasm and Flye. Therefore, I created Mabs 2 that uses Hifiasm and Flye. Mabs 1 worked in a 4-dimensional parameter space (optimized 4 different parameters of Miniasm), while Mabs 2 works in a 1-dimensional parameter space.
+b) "Busco Score" is because very early versions of Mabs simply maximized BUSCO's "S" (the number of single copy genes). However, I quickly realized that maximization of S may lead to collapsing of close paralogs, because it transfers them from the "multicopy" category to the "single-copy" category, thus increasing S. To deal with this problem, I started to classify multicopy genes into true multicopy (TM) and false multicopy (FM), and devised AG as a target for maximization, which is a sum of S and TM.
+10. How to cite Mabs?
+a link to BioRxiv will be here in a few days .
+