[jlazovskis] This document contains information about who will do which jobs / run which experiments. Also information on what to do differently.
[jlazovskis] This is about redoing the Frontiers analysis on a larger circuit, either the whole somatosensory cortex, or a cylinder of it.
-
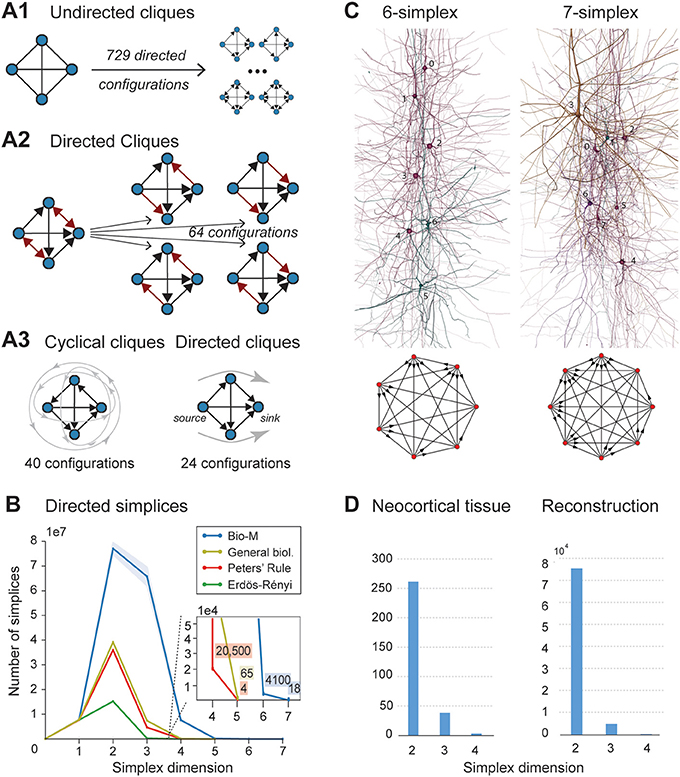
- [Henri] I'll contact Michael concerning the general biology and Peter's rule and try to work on Figure 2B.
- [Henri] So Michael replied that Peter's rule is not feasible, so I just do BioM, ER and general bio.
-
- A1 - A3 will be different, as now L2 and L3 are separate.
- [jlazovskis] Redone here, code in
proj102/lazovski/frontiers-figs-redo/5_make-visuals.py. - [jlazovskis] Variants of subfigures are in the
figfolder, namedfig3-XX.png. Code is in same folder for those.
-
- E is very interesting
-
- Something like C would be great
-
Figure S5
- Similar to Figure 6E, but in more detail
-
Figure S8
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
-
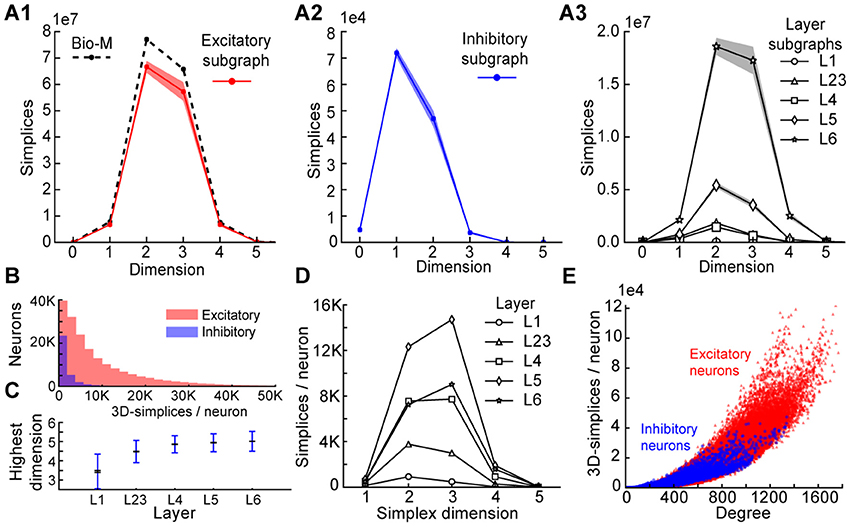
[JasonPSmith] For each neuron G we can compute the number of simplices for which G is a sink/source, we can then look at the distribution of sink/source neurons by type.
- [seirios] Cool! Definitely, doing analyses at the level of cell type is the next step!
[Nicolas] I'll try to details for each proposed experiments what I think the goals are, what needs to be done, how we can do it, and what I(we) might expect. For clarity it may be easier to give each experiments his own file.
We soon(ish) will have access to three S1v6 somatosensory circuit each based on the same rat statistics but with different seeds (as opposed to frontier where we had five different rat and an average one).
[seirios] The variation between the three builds is in the cell densities, and in the random seeds that go into every part of circuit building (cell placement, morphology selection, sampling of touches, etc.).
Such a circuit was already created a year ago, those three have seen some improvment (in what?) from the earlier version.
[seirios] The new circuits are built using the latest pipeline, with a better atlas and an updated connection recipe.
To have some numbers in mind here is the results of the counting of simplices of this earlier circuit that Gard did a year ago.
- Dim 0: 1722604 (~1.7 million)
- Dim 1: 1059736608 (~1.1 billion)
- Dim 2: 22705492972 (~22 billion)
- Dim 3: 58421988203 (~58 billion)
- Dim 4: 30838397649 (~31 billion)
- Dim 5: 5082042041 (~5.1 billion)
- Dim 6: 337370565 (~0.33 billion)
- Dim 7: 9360457 (~9.4 million)
- Dim 8: 108025 (~0.1 million)
- Dim 9: 458
We probably cannot compute any homology on such a big complexe, but possibly on smaller subgraph.
[seirios] I have a first version of the central region (one microcolumn's width away from the border), and it's 1.22M cells, so a bit more manageable than the whole circuit (~1.7M).
The experiments we want to do can be divided roughly into the one who needs activity and the one who only looks at the circuit (structural). Obviously we will use structural results also with activity. So first lets descibe the easy structural computation and their variation
Goal:
The goal here is to redo the computation Gard Did on the three new circuit. We then compare those number to other more or less random graph with similar properties (ER with same number of vertex and edges for example).
How to do it: This is pretty straightforward with flagsercount, but it can takes time. Also storing simplices might not be possible (at least not all of them) something that would often be nice. One possible way to get around this is to only store the highest dimension, to store a random subset, or to store simplices for smaller (meaningfull) subgraph, whatever meaningfull would mean.
[seirios] According to Jason's estimate, it would take 0.5TB to store all simplices. That sounds perfectly doable!
[JasonPSmith] Note that the 0.5TB is very much a lower bound, I expect it is higher, how much higher depends on how clever we are about storing it. Also on run times, to compute the simplex counts on the previous somatosensory circuit took 7.5hrs, using 256 cores, and used 55GB of memory.
[JasonPSmith] I have adapted flagser to print the simplices in a compressed way. So each simplex is stored as at most 4 x 64 bit ints. The output has a size of 1.6TB.
[seirios] Just for fun, I implemented independently the simplex count and got the exact same results as shown above! Total runtime was 50 min (33 min for the longest job), splitting the total number of cells among 18 jobs (max 100000 cells per job) of one node each, using 72 cores and 285 GB of RAM per node.
Variations:
Here is a non comprehensive list of variation around this, some may be stupid/useless:
- Divide the big circuit into columns then compare them or compare to v5
- Grow the circuit from a small one to the complete one (maybe start with a cylinder to keep all layers then increase the radius)
- On the same idea, I think Henry M. asked what is the smallest region with highest dimensional simplices (this would be the smallest volume of the highest dimensional simplices but I think he meant more on something related to the previous item)
- Harder but may be possible, count other motifs (cliques or tournaments), all motifs on 3 vertices [JasonPSmith] Counting tournaments should be easy and I think is worth doing. I have a variation of flagser-count that does this.
- [JasonPSmith] Kathryn recommended looking at pathways, in particular from L4 excitatory to L2/3 excitatory. Do we get more 4 at source and L2/3 at sink?
Figure 3 give us some other things we can do
- divide by excitatory/inhibitory
- by layers
[seirios] More generally, divide by morphological type, as that comprises both layer and excitatory/inhibitory and allows studying pathways (e.g. connections between some class of interneurons and pyramidal cells), which are biologically relevant.
[JasonPSmith] We might be able to do some homology computations. If we restrict to a column, we might be able to compute homology in some dimensions, as in frontiers.
Goal:
Understand simplices with respect to usual descriptor of the neuron, here we are still looking at thing without any activity.
How to do it: With the list of simplices its elementary to do. What takes time is to analyze and trying to makes sense of what you see. There is a ton of variation so its easy to get lost.
Variations: So the goal is to look at things like information given by simplices vs information of the neuron.
For example : "highest dimension a neuron is part of" vs "EXC/INH" ( or mtype or layer) etc.
Here are a few neuronal information you can get from the listing of the simplices (in fig3 for instance):
- the highest dimension of a simplice a neuron is part of
- number of n-simplices a neuron belongs to
jason proposal:
- number of simplices a neuron is a sink or source
We can also look at population and not only at a neuron level, ie fig3.C.
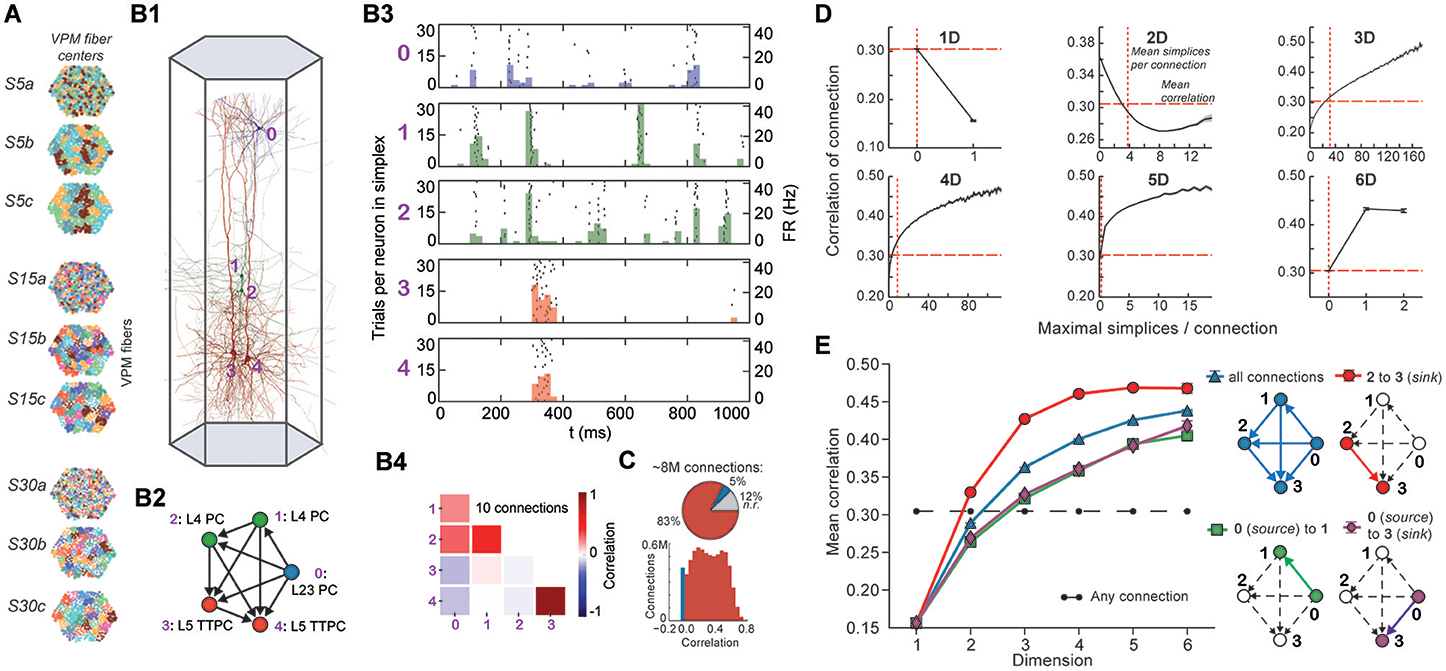
Like we discussed on the meeting of october the 29th, we need to define stimuli. This is probably something that would need to be discussed in the next meeting with Henry M. and maybe before with Michael.
I think that in the context of following frontiers protocol, we should take the same stimuli (but this might be a bad idea for many reason)
In any case this stimuli were designed for one column, so even if we settled on using this, how do we apply this to the whole SSCx?
[seirios] There are thalamic fibers all over the place, so in principle we have only to feed the spike trains through the fibers.
[JasonPSmith] Michael said using the same stimuli as frontiers is the best approach.
One idea was to activate only one column (so very close to frontiers). At the opposite there is giving the stimuli to the whole network and every combination in between. Also even this is not well defined for me, is the circuit strictly divided into column to which we can give the same stimulus or is it more blurry?
[seirios] The circuit is one big mass of cells and is not divided into columns. However, we can define columns at any location and of any size.
[JasonPSmith] Michael said selecteding a mc2 sized column from the centre of the region would be best. But perhaps to also run it on the whole circuit and or larger columns as well.
[seirios] Regarding the runtime of the simulations. For the whole circuit it's about 13h and for a column it's between 1-2h (depending on its size). This is for 4 seconds of biological time.
For now lets suppose we have some stimuli given to the circuit, and describes the experiments:
Goal:
compute pearson correlation between any pairs of neuron. This would be a preliminary steps to a lot of things.
How to do it:
I'm not sure how much time it'd take to compute those though. I think computing correlation on 30K took a few minutes so on 1.7M it may be 50^2 times longer so at least a week. Obviously we can use many nodes (I'm also not sure if the computation was using all the core It was a scipy thing so I would say no). Overall if we are efficient with parallelization it should actually be pretty fast (250 cpu per node should make this takes a few hours top).
[JasonPSmith] From the meeting: We can restrict to pairs that are connected to each other.
[seirios] For maximum efficiency, I'd vouch for custom C code, which I volunteer to write. We'd only have to define exactly how we are computing the correlations.
Variations: Its not always clear what data we want to compute correlation on. By this I mean we can have many correlation for each simulation, or we can concatenate those( what they did in frontier) to get only one number per pair of neuron.
Once we have correlations and simplices computed we can do:
- Figure 4 E, ie correlation in a simplex depeding on respective position
- S5 same as 4E with variation
- also other in fig 4
- S8 but I dont understand fully this one, we should ask Micheal how everythin is defined
{kind=link}
{kind=link}
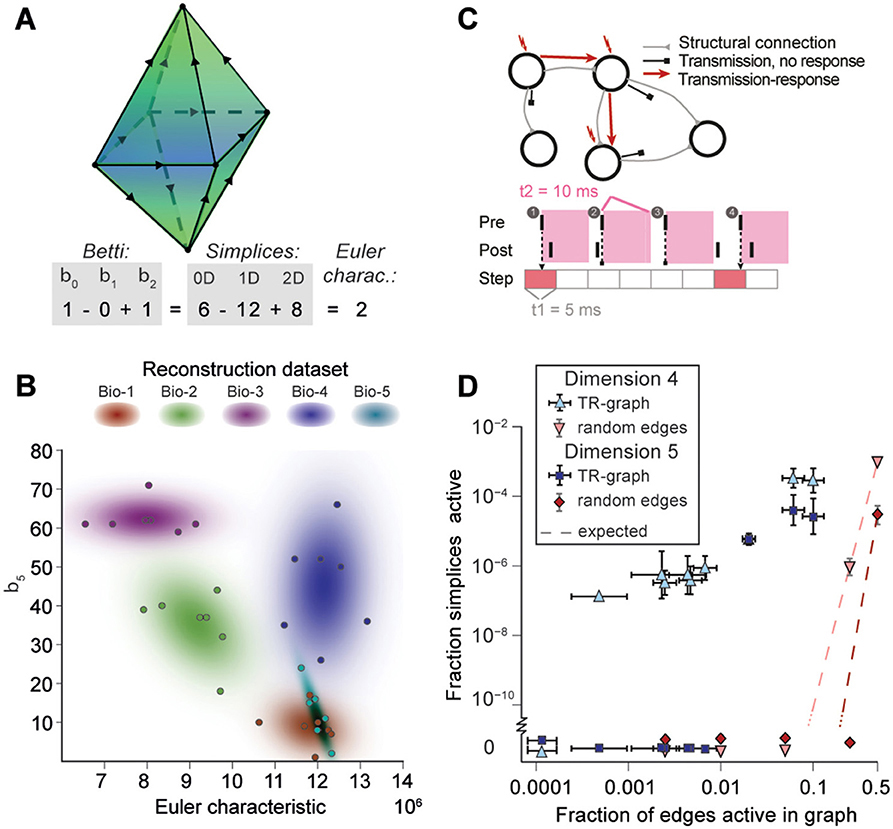
Goal:
Compute the TR graphs and their topology
How to do it: Micheal's code, we should probably take frontiers parameter. Its not clear atm wether or not we will be able to compute homology of those TR.
Once TR computed we can do:
- Figure 6C doesnt necesseraly require 6B,
- Figure 6A,B,D require betti computation but we could try just with cell count or EC
It might be interesting to look at those.