In this example ComplexMixtures.jl is used to study the interactions of a POPC membrane with a mixture of 20%(mol/mol) ethanol in water. At this concentration ethanol destabilizes the membrane.
+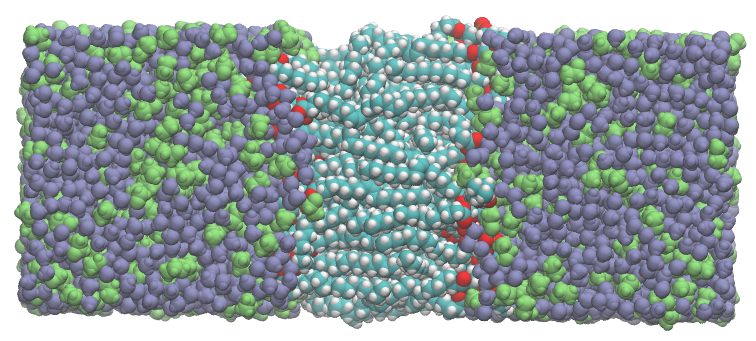 +
+System image: a POPC membrane (center) solvated by a mixture of water (purple) and ethanol (green). The system is composed by 59 POPC, 5000 water, and 1000 ethanol molecules.
The files required to run this example are:
- equilibrated.pdb: The PDB file of the complete system.
- traj_POPC.dcd: Trajectory file. This is a 365Mb file, necessary for running from scratch the calculations.
To run the scripts, we suggest the following procedure:
- Create a directory, for example
example3. - Copy the required data files above to this directory.
- Launch
julia in that directory: activate the directory environment, and install the required packages. This launching Julia and executing:import Pkg
+Pkg.activate(".")
+Pkg.add(["ComplexMixtures", "PDBTools", "Plots", "LaTeXStrings", "EasyFit"])
+exit()
- Copy the code of each script in to a file, and execute with:
julia -t auto script.jl
Alternativelly (and perhaps preferrably), copy line by line the content of the script into the Julia REPL, to follow each step of the calculation.
Here we show the distribution functions and KB integrals associated to the solvation of the membrane by water and ethanol.
Complete example code: click here!
import Pkg;
+Pkg.activate(".");
+
+using PDBTools
+using ComplexMixtures
+using Plots
+using LaTeXStrings
+using EasyFit: movavg
+
+# The full trajectory file is available at:
+# https://www.dropbox.com/scl/fi/hcenxrdf8g8hfbllyakhy/traj_POPC.dcd?rlkey=h9zivtwgya3ivva1i6q6xmr2p&dl=0
+trajectory_file = "./traj_POPC.dcd"
+
+# Load a PDB file of the system
+system = readPDB("./equilibrated.pdb")
+
+# Select the atoms corresponding to glycerol and water
+popc = select(system, "resname POPC")
+water = select(system, "water")
+ethanol = select(system, "resname ETOH")
+
+# Set the complete membrane as the solute. We use nmols=1 here such
+# that the membrane is considered a single solute in the calculation.
+solute = AtomSelection(popc, nmols=1)
+
+# Compute water-POPC distribution and KB integral
+solvent = AtomSelection(water, natomspermol=3)
+
+# Set the trajectory structure
+trajectory = Trajectory(trajectory_file, solute, solvent)
+
+# We want to get reasonably converged KB integrals, which usually
+# require large solute domains. Distribution functions converge
+# rapidly (~10Angs or less), on the other side.
+options = Options(bulk_range=(15.0, 20.0))
+
+# Compute the mddf and associated properties
+mddf_water_POPC = mddf(trajectory, options)
+
+# Save results to file for later use
+save(mddf_water_POPC, "./mddf_water_POPC.json")
+println("Results saved to ./mddf_water_POPC.json file")
+
+# Compute ethanol-POPC distribution and KB integral
+solvent = AtomSelection(ethanol, natomspermol=9)
+traj = Trajectory(trajectory_file, solute, solvent)
+mddf_ethanol_POPC = mddf(traj, options)
+
+# Save results for later use
+save(mddf_ethanol_POPC, "./mddf_ethanol_POPC.json")
+println("Results saved to ./mddf_ethanol_POPC.json file")
+
+#
+# Plot the MDDF and KB integrals
+#
+# Plot defaults
+plot_font = "Computer Modern"
+default(
+ fontfamily=plot_font,
+ linewidth=2.5,
+ framestyle=:box,
+ label=nothing,
+ grid=false,
+ palette=:tab10
+)
+scalefontsizes();
+scalefontsizes(1.3);
+
+#
+# Plots cossolvent-POPC MDDFs in subplot 1
+#
+plot(layout=(2, 1))
+# Water MDDF
+plot!(
+ mddf_water_POPC.d, # distances
+ movavg(mddf_water_POPC.mddf, n=10).x, # water MDDF - smoothed
+ label="Water",
+ subplot=1
+)
+# Ethanol MDDF
+plot!(
+ mddf_ethanol_POPC.d, # distances
+ movavg(mddf_ethanol_POPC.mddf, n=10).x, # water MDDF - smoothed
+ label="Ethanol",
+ subplot=1
+)
+# Plot settings
+plot!(
+ xlabel=L"\textrm{Distance / \AA}",
+ ylabel="MDDF",
+ xlim=(0, 10),
+ subplot=1
+)
+
+#
+# Plot cossolvent-POPC KB integrals in subplot 2
+#
+# Water KB
+plot!(
+ mddf_water_POPC.d, # distances
+ mddf_water_POPC.kb, # water KB
+ label="Water",
+ subplot=2
+)
+# Ethanol KB
+plot!(
+ mddf_ethanol_POPC.d, # distances
+ mddf_ethanol_POPC.kb, # ethanol KB
+ label="Ethanol",
+ subplot=2
+)
+# Plot settings
+plot!(
+ xlabel=L"\textrm{Distance / \AA}",
+ ylabel=L"\textrm{KB~/~L~mol^{-1}}",
+ xlim=(0, 10),
+ subplot=2
+)
+
+savefig("popc_water_ethanol_mddf_kb.png")
+println("Plot saved to popc_water_ethanol_mddf_kb.png file")
+
+
+
The distribution functions are shown in the first panel of the figure below, and the KB integrals are shown in the second panel.

Clearly, both water and ethanol accumulate on the proximity of the membrane. The distribution functions suggest that ethanol displays a greater local density augmentation, reaching concentrations roughly 4 times higher than bulk concentrations. Water has a peak at hydrogen-bonding distances (~1.8$\mathrm{\AA}$) and a secondary peak at 2.5$\mathrm{\AA}$.
Despite the fact that ethanol displays a greater relative density (relative to its own bulk concentration) at short distances, the KB integral of water turns out to be greater (more positive) than that of ethanol. This implies that the membrane is preferentially hydrated.
The minimum-distance distribution function can be decomposed into the contributions of the ethanol molecule groups.
Complete example code: click here!
import Pkg;
+Pkg.activate(".");
+
+using ComplexMixtures
+using PDBTools
+using Plots
+using LaTeXStrings
+using EasyFit: movavg
+
+# Some default settings for the plots
+plot_font = "Computer Modern"
+Plots.default(
+ fontfamily=plot_font,
+ linewidth=1.5,
+ framestyle=:box,
+ label=nothing,
+ grid=false,
+)
+scalefontsizes();
+scalefontsizes(1.3);
+
+# Read system PDB file
+system = readPDB("equilibrated.pdb")
+ethanol = select(system, "resname ETOH")
+
+# Load the pre-calculated MDDF of ethanol
+mddf_ethanol_POPC = load("mddf_ethanol_POPC.json")
+
+#
+# Contributions of the ethanol groups
+#
+# Define the groups using selections. Set a dict, in which the keys are the group names
+# and the values are the selections
+groups = (
+ "Hydroxyl" => select(ethanol, "name O1 or name HO1"),
+ "Aliphatic chain" => select(ethanol, "not name O1 and not name HO1"),
+)
+# plot the total mddf and the contributions of the groups
+x = mddf_ethanol_POPC.d
+plot(x, movavg(mddf_ethanol_POPC.mddf, n=10).x, label="Total MDDF")
+for (group_name, group_atoms) in groups
+ cont = contributions(mddf_ethanol_POPC, SolventGroup(group_atoms))
+ y = movavg(cont, n=10).x
+ plot!(x, y, label=group_name)
+end
+# Plot settings
+plot!(
+ xlim=(1, 8),
+ xlabel=L"\textrm{Distance / \AA}",
+ ylabel="MDDF"
+)
+savefig("./mddf_ethanol_groups.png")
+println("The plot was saved as mddf_ethanol_groups.png")
In the figure below we show the contributions of the ethanol hydroxyl and aliphatic chain groups to the total MDDF.

As expected, the MDDF at hydrogen-bonding distances is composed by contributions of the ethanol hydroxyl group, and the non-specific interactions at ~2.5$\mathrm{\AA}$ have a greater contribution of the aliphatic chain of the solvent molecules. It is interesting to explore the chemical complexity of POPC in what concerns these interactions.
The MDDF can also be decomposed into the contributions of the solute atoms and chemical groups. First, we show the contributions of the POPC chemical groups to the water-POPC distribution.
Complete example code: click here!
import Pkg;
+Pkg.activate(".");
+
+using ComplexMixtures
+using PDBTools
+using Plots
+using LaTeXStrings
+using EasyFit: movavg
+
+# Some default settings for the plots
+plot_font = "Computer Modern"
+Plots.default(
+ fontfamily=plot_font,
+ linewidth=2,
+ framestyle=:box,
+ label=nothing,
+ grid=false,
+)
+scalefontsizes();
+scalefontsizes(1.3);
+
+# Read system PDB file
+system = readPDB("equilibrated.pdb")
+
+# Load the pre-calculated MDDF of water
+mddf_water_POPC = load("mddf_water_POPC.json")
+
+#
+# Here we define the POPC groups, from the atom names. Each group
+# is a vector of atom names, and the keys are the group names.
+#
+groups = (
+ "Choline" => ["N", "C12", "H12A", "H12B", "C13", "H13A", "H13B", "H13C", "C14",
+ "H14A", "H14B", "H14C", "C15", "H15A", "H15B", "H15C", "C11", "H11A", "H11B"],
+ "Phosphate" => ["P", "O13", "O14", "O12"],
+ "Glycerol" => ["O11", "C1", "HA", "HB", "C2", "HS", "O21", "C3", "HX", "HY", "O31"],
+ "Oleoyl" => ["O22", "C21", "H2R", "H2S", "C22", "C23", "H3R", "H3S", "C24", "H4R", "H4S",
+ "C25", "H5R", "H5S", "C26", "H6R", "H6S", "C27", "H7R", "H7S", "C28", "H8R", "H8S",
+ "C29", "H91", "C210", "H101", "C211", "H11R", "H11S", "C212", "H12R", "H12S",
+ "C213", "H13R", "H13S", "C214", "H14R", "H14S", "C215", "H15R", "H15S",
+ "C216", "H16R", "H16S", "C217", "H17R", "H17S", "C218", "H18R", "H18S", "H18T"],
+ "Palmitoyl" => ["C31", "O32", "C32", "H2X", "H2Y", "C33", "H3X", "H3Y", "C34", "H4X", "H4Y",
+ "C35", "H5X", "H5Y", "C36", "H6X", "H6Y", "C37", "H7X", "H7Y", "C38", "H8X",
+ "H8Y", "C39", "H9X", "H9Y", "C310", "H10X", "H10Y", "C311", "H11X", "H11Y",
+ "C312", "H12X", "H12Y", "C313", "H13X", "H13Y", "C314", "H14X", "H14Y", "C315",
+ "H15X", "H15Y", "C316", "H16X", "H16Y", "H16Z"],
+)
+
+#
+# plot the total mddf and the contributions of the groups
+#
+x = mddf_water_POPC.d
+plot(
+ x,
+ movavg(mddf_water_POPC.mddf, n=10).x,
+ label="Total water-POPC MDDF"
+)
+for (group_name, group_atoms) in groups
+ cont = contributions(mddf_water_POPC, SoluteGroup(group_atoms))
+ y = movavg(cont, n=10).x
+ plot!(x, y, label=group_name)
+end
+# Plot settings
+plot!(
+ xlim=(1, 5),
+ xlabel=L"\textrm{Distance / \AA}",
+ ylabel="MDDF"
+)
+savefig("./mddf_POPC_water_groups.png")
+println("The plot was saved as mddf_POPC_water_groups.png")
+
+

Not surprisingly, water interactions occur majoritarily with the Phosphate and Choline groups of POPC molecules, that is, with the polar head of the lipid. The interactions at hydrogen-bonding distances are dominated by the phosphate group, and non-specific interaction occur mostly with the choline group. Some water molecules penetrate the membrane and interact with the glycerol and aliphatic chains of POPC, but these contributions are clearly secondary.
The interactions of ethanol molecules with the membrane are more interesting, because ethanol penetrates the membrane. Here we decompose the ethanol-POPC distribution function into the contributions of the POPC chemical groups.
Complete example code: click here!
import Pkg;
+Pkg.activate(".");
+
+using ComplexMixtures
+using PDBTools
+using Plots
+using LaTeXStrings
+using EasyFit: movavg
+
+# Some default settings for the plots
+plot_font = "Computer Modern"
+Plots.default(
+ fontfamily=plot_font,
+ linewidth=2,
+ framestyle=:box,
+ label=nothing,
+ grid=false,
+)
+scalefontsizes();
+scalefontsizes(1.3);
+
+# Read system PDB file
+system = readPDB("equilibrated.pdb")
+
+# Load the pre-calculated MDDF of ethanol
+mddf_ethanol_POPC = load("mddf_ethanol_POPC.json")
+
+#
+# Here we define the POPC groups, from the atom names. Each group
+# is a vector of atom names, and the keys are the group names.
+#
+groups = (
+ "Choline" => ["N", "C12", "H12A", "H12B", "C13", "H13A", "H13B", "H13C", "C14",
+ "H14A", "H14B", "H14C", "C15", "H15A", "H15B", "H15C", "C11", "H11A", "H11B"],
+ "Phosphate" => ["P", "O13", "O14", "O12"],
+ "Glycerol" => ["O11", "C1", "HA", "HB", "C2", "HS", "O21", "C3", "HX", "HY", "O31"],
+ "Oleoyl" => ["O22", "C21", "H2R", "H2S", "C22", "C23", "H3R", "H3S", "C24", "H4R", "H4S",
+ "C25", "H5R", "H5S", "C26", "H6R", "H6S", "C27", "H7R", "H7S", "C28", "H8R", "H8S",
+ "C29", "H91", "C210", "H101", "C211", "H11R", "H11S", "C212", "H12R", "H12S",
+ "C213", "H13R", "H13S", "C214", "H14R", "H14S", "C215", "H15R", "H15S",
+ "C216", "H16R", "H16S", "C217", "H17R", "H17S", "C218", "H18R", "H18S", "H18T"],
+ "Palmitoyl" => ["C31", "O32", "C32", "H2X", "H2Y", "C33", "H3X", "H3Y", "C34", "H4X", "H4Y",
+ "C35", "H5X", "H5Y", "C36", "H6X", "H6Y", "C37", "H7X", "H7Y", "C38", "H8X",
+ "H8Y", "C39", "H9X", "H9Y", "C310", "H10X", "H10Y", "C311", "H11X", "H11Y",
+ "C312", "H12X", "H12Y", "C313", "H13X", "H13Y", "C314", "H14X", "H14Y", "C315",
+ "H15X", "H15Y", "C316", "H16X", "H16Y", "H16Z"],
+)
+
+#
+# plot the total mddf and the contributions of the groups
+#
+x = mddf_ethanol_POPC.d
+plot(
+ x,
+ movavg(mddf_ethanol_POPC.mddf, n=10).x,
+ label="Total ethanol-POPC MDDF"
+)
+for (group_name, group_atoms) in groups
+ cont = contributions(mddf_ethanol_POPC, SoluteGroup(group_atoms))
+ y = movavg(cont, n=10).x
+ plot!(x, y, label=group_name)
+end
+# Plot settings
+plot!(
+ xlim=(1.3, 5),
+ ylim=(0, 1.8),
+ xlabel=L"\textrm{Distance / \AA}",
+ ylabel="MDDF"
+)
+savefig("./mddf_POPC_ethanol_groups.png")
+println("The plot was saved as mddf_POPC_ethanol_groups.png")
+

Ethanol molecules interact with the choline and phosphate groups of POPC molecules, as do water molecules. The contributions to the MDDF at hydrogen-bonding distances come essentially from ethanol-phosphate interactions.
However, ethanol molecules interact frequently with the glycerol and aliphatic chains of POPC. Interactions with the Oleoyl chain are slightly stronger than with the Palmitoyl chain. This means that ethanol penetrates the hydrophobic core of the membrane, displaying non-specific interactions with the lipids and with the glycerol group. These interactions are probably associated to the destabilizing role of ethanol in the membrane structure.
The MDDFs can be decomposed at more granular level, in which each chemical group of the aliphatic chains of the POPC molecules are considered independently. This allows the study of the penetration of the ethanol molecules in the membrane. In the figure below, the carbonyl following the glycerol group of the POPC molecules is represented in the left, and going to the right the aliphatic chain groups are sequentially shown.
Complete example code: click here!
import Pkg;
+Pkg.activate(".");
+
+using ComplexMixtures
+using PDBTools
+using Plots
+using LaTeXStrings
+using EasyFit: movavg
+
+# Some default settings for the plots
+plot_font = "Computer Modern"
+Plots.default(
+ fontfamily=plot_font,
+ linewidth=2,
+ framestyle=:box,
+ label=nothing,
+ grid=false,
+)
+scalefontsizes();
+scalefontsizes(1.3);
+
+# Read system PDB file
+system = readPDB("equilibrated.pdb")
+
+# Load the pre-calculated MDDF of ethanol
+mddf_ethanol_POPC = load("mddf_ethanol_POPC.json")
+
+# Splitting the oleoyl chain into groups along the the chain.
+# The labels `CH_2` etc stand for `CH₂`, for example, in LaTeX notation,
+# for a nicer plot axis ticks formatting.
+oleoyl_groups = (
+ "CO" => ["O22", "C21"],
+ "CH_2" => ["H2R", "H2S", "C22"],
+ "CH_2" => ["C23", "H3R", "H3S"],
+ "CH_2" => ["C24", "H4R", "H4S"],
+ "CH_2" => ["C25", "H5R", "H5S"],
+ "CH_2" => ["C26", "H6R", "H6S"],
+ "CH_2" => ["C27", "H7R", "H7S"],
+ "CH_2" => ["C28", "H8R", "H8S"],
+ "CH" => ["C29", "H91"],
+ "CH" => ["C210", "H101"],
+ "CH_2" => ["C211", "H11R", "H11S"],
+ "CH_2" => ["C212", "H12R", "H12S"],
+ "CH_2" => ["C213", "H13R", "H13S"],
+ "CH_2" => ["C214", "H14R", "H14S"],
+ "CH_2" => ["C215", "H15R", "H15S"],
+ "CH_2" => ["C216", "H16R", "H16S"],
+ "CH_2" => ["C217", "H17R", "H17S"],
+ "CH_3" => ["C218", "H18R", "H18S", "H18T"]
+)
+
+# Format tick labels with LaTeX
+labels_o = [latexstring("\\textrm{$key}") for (key, val) in oleoyl_groups]
+
+# We first collect the contributions of each group into a vector of vectors:
+gcontrib = Vector{Float64}[] # empty vector of vectors
+for (group_name, group_atoms) in oleoyl_groups
+ group_contributions = contributions(mddf_ethanol_POPC, SoluteGroup(group_atoms))
+ push!(gcontrib, movavg(group_contributions; n=10).x)
+end
+
+# Convert the vector of vectors into a matrix
+gcontrib = stack(gcontrib)
+
+# Find the indices of the MDDF where the distances are between 1.5 and 3.0 Å
+idmin = findfirst(d -> d > 1.5, mddf_ethanol_POPC.d)
+idmax = findfirst(d -> d > 3.0, mddf_ethanol_POPC.d)
+
+# The plot will have two lines, the first plot will contain the
+# oleoyl groups contributions, and the second plot will contain the
+# contributions of the palmitoyl groups.
+plot(layout=(2, 1))
+
+# Plot the contributions of the oleoyl groups
+contourf!(
+ 1:length(oleoyl_groups),
+ mddf_ethanol_POPC.d[idmin:idmax],
+ gcontrib[idmin:idmax, :],
+ color=cgrad(:tempo), linewidth=1, linecolor=:black,
+ colorbar=:none, levels=10,
+ ylabel=L"r/\AA", xrotation=60,
+ xticks=(1:length(oleoyl_groups), labels_o),
+ subplot=1,
+)
+annotate!(14, 2.7, text("Oleoyl", :left, 12, plot_font), subplot=1)
+
+#
+# Repeat procedure for the palmitoyl groups
+#
+palmitoyl_groups = (
+ "CO" => ["C31", "O32"],
+ "CH_2" => ["C32", "H2X", "H2Y"],
+ "CH_2" => ["C33", "H3X", "H3Y"],
+ "CH_2" => ["C34", "H4X", "H4Y"],
+ "CH_2" => ["C35", "H5X", "H5Y"],
+ "CH_2" => ["C36", "H6X", "H6Y"],
+ "CH_2" => ["C37", "H7X", "H7Y"],
+ "CH_2" => ["C38", "H8X", "H8Y"],
+ "CH_2" => ["C39", "H9X", "H9Y"],
+ "CH_2" => ["C310", "H10X", "H10Y"],
+ "CH_2" => ["C311", "H11X", "H11Y"],
+ "CH_2" => ["C312", "H12X", "H12Y"],
+ "CH_2" => ["C313", "H13X", "H13Y"],
+ "CH_2" => ["C314", "H14X", "H14Y"],
+ "CH_2" => ["C315", "H15X", "H15Y"],
+ "CH_3" => ["C316", "H16X", "H16Y", "H16Z"],
+)
+
+# Format tick labels with LaTeX
+labels_p = [latexstring("\\textrm{$key}") for (key, val) in palmitoyl_groups]
+
+# We first collect the contributions of each group into a # vector of vectors:
+gcontrib = Vector{Float64}[] # empty vector of vectors
+for (group_name, group_atoms) in palmitoyl_groups
+ group_contributions = contributions(mddf_ethanol_POPC, SoluteGroup(group_atoms))
+ push!(gcontrib, movavg(group_contributions; n=10).x)
+end
+
+# Convert the vector of vectors into a matrix
+gcontrib = stack(gcontrib)
+
+# Plot the contributions of the palmitoyl groups
+contourf!(
+ 1:length(palmitoyl_groups),
+ mddf_ethanol_POPC.d[idmin:idmax],
+ gcontrib[idmin:idmax, :],
+ color=cgrad(:tempo), linewidth=1, linecolor=:black,
+ colorbar=:none, levels=10,
+ xlabel="Group",
+ ylabel=L"r/\AA", xrotation=60,
+ xticks=(1:length(labels_p), labels_p),
+ bottom_margin=0.5Plots.Measures.cm,
+ subplot=2,
+)
+annotate!(12, 2.7, text("Palmitoyl", :left, 12, plot_font), subplot=2)
+
+savefig("POPC_ethanol_chains.png")
+println("The plot was saved as POPC_ethanol_chains.png")
+
+#
+# Now, plot a similar map for the water interactions with the POPC chain
+#
+mddf_water_POPC = load("mddf_water_POPC.json")
+plot(layout=(2, 1))
+
+# Plot the contributions of the oleoyl groups
+# We first collect the contributions of each group into a vector of vectors:
+gcontrib = Vector{Float64}[] # empty vector of vectors
+for (group_name, group_atoms) in oleoyl_groups
+ group_contributions = contributions(mddf_water_POPC, SoluteGroup(group_atoms))
+ push!(gcontrib, movavg(group_contributions; n=10).x)
+end
+# Convert the vector of vectors into a matrix
+gcontrib = stack(gcontrib)
+# Plot matrix as density map
+contourf!(
+ 1:length(oleoyl_groups),
+ mddf_water_POPC.d[idmin:idmax],
+ gcontrib[idmin:idmax, :],
+ color=cgrad(:tempo), linewidth=1, linecolor=:black,
+ colorbar=:none, levels=10,
+ ylabel=L"r/\AA", xrotation=60,
+ xticks=(1:length(oleoyl_groups), labels_o), subplot=1,
+)
+annotate!(14, 2.7, text("Oleoyl", :left, 12, plot_font), subplot=1)
+
+# Plot the contributions of the palmitoyl groups
+# We first collect the contributions of each group into a vector of vectors:
+gcontrib = Vector{Float64}[] # empty vector of vectors
+for (group_name, group_atoms) in palmitoyl_groups
+ group_contributions = contributions(mddf_water_POPC, SoluteGroup(group_atoms))
+ push!(gcontrib, movavg(group_contributions; n=10).x)
+end
+# Convert the vector of vectors into a matrix
+gcontrib = stack(gcontrib)
+# Plot matrix as density map
+contourf!(
+ 1:length(palmitoyl_groups),
+ mddf_water_POPC.d[idmin:idmax],
+ gcontrib[idmin:idmax, :],
+ color=cgrad(:tempo), linewidth=1, linecolor=:black,
+ colorbar=:none, levels=10,
+ xlabel="Group",
+ ylabel=L"r/\AA", xrotation=60,
+ xticks=(1:length(palmitoyl_groups), labels_o),
+ subplot=2,
+ bottom_margin=0.5Plots.Measures.cm,
+)
+annotate!(12, 2.7, text("Palmitoyl", :left, 12, plot_font), subplot=2)
+
+savefig("POPC_water_chains.png")
+println("The plot was saved as POPC_water_chains.png")

Ethanol displays an important density augmentation at the vicinity of the carbonyl that follows the glycerol group, and accumulates on the proximity of the aliphatic chain. The density of ethanol decreases as one advances into the aliphatic chain, displaying a minimum around the insaturation in the Oleoyl chain. The terminal methyl group of both chains display a greater solvation by ethanol, suggesting the twisting of the aliphatic chain expose these terminal groups to membrane depth where ethanol is already abundant.
The equivalent maps for water are strikingly different, and show that water is excluded from the interior of the membrane:

Membrane built with the VMD membrane plugin.
Water and ethanol layers added with Packmol.
The simulations were performed with NAMD, with CHARMM36 parameters.
Density of the ethanol-water mixture from: https://wissen.science-and-fun.de/chemistry/chemistry/density-tables/ethanol-water-mixtures/


 +
+ +
+ +
+ +
+ +
+ +
+


 +
+ +
+


 +
+ +
+ +
+ +
+

 +
+ +
+ +
+ ","category":"page"},{"location":"results/","page":"Results","title":"Results","text":"Thus, this plot was obtained with the following code:","category":"page"},{"location":"results/","page":"Results","title":"Results","text":"using Plots\nplot(results.d,results.mddf,xlabel=\"d/A\",ylabel=\"mddf(d) / L/mol\") ","category":"page"},{"location":"results/#Kirkwood-Buff-integral:-results.kb","page":"Results","title":"Kirkwood-Buff integral: results.kb","text":"","category":"section"},{"location":"results/","page":"Results","title":"Results","text":"The results.kb vector will contain the Kirkwood-Buff integral computed as a function of the minimum-distance to the solute. For properly sampled simulations, it is expected to converge at large distances. ","category":"page"},{"location":"results/","page":"Results","title":"Results","text":"julia> results.kb\n500-element Array{Float64,1}:\n 0.0\n -0.3249356504752985\n -2.9804719721525\n ⋮\n 0.72186381783\n 1.13624162115","category":"page"},{"location":"results/","page":"Results","title":"Results","text":"A typical plot of results.kb as a function of results.d will look like:","category":"page"},{"location":"results/","page":"Results","title":"Results","text":"
","category":"page"},{"location":"results/","page":"Results","title":"Results","text":"Thus, this plot was obtained with the following code:","category":"page"},{"location":"results/","page":"Results","title":"Results","text":"using Plots\nplot(results.d,results.mddf,xlabel=\"d/A\",ylabel=\"mddf(d) / L/mol\") ","category":"page"},{"location":"results/#Kirkwood-Buff-integral:-results.kb","page":"Results","title":"Kirkwood-Buff integral: results.kb","text":"","category":"section"},{"location":"results/","page":"Results","title":"Results","text":"The results.kb vector will contain the Kirkwood-Buff integral computed as a function of the minimum-distance to the solute. For properly sampled simulations, it is expected to converge at large distances. ","category":"page"},{"location":"results/","page":"Results","title":"Results","text":"julia> results.kb\n500-element Array{Float64,1}:\n 0.0\n -0.3249356504752985\n -2.9804719721525\n ⋮\n 0.72186381783\n 1.13624162115","category":"page"},{"location":"results/","page":"Results","title":"Results","text":"A typical plot of results.kb as a function of results.d will look like:","category":"page"},{"location":"results/","page":"Results","title":"Results","text":" ","category":"page"},{"location":"results/","page":"Results","title":"Results","text":"Thus, this plot was obtained with the following code:","category":"page"},{"location":"results/","page":"Results","title":"Results","text":"using Plots\nplot(results.d,results.kb,xlabel=\"d/A\",ylabel=\"mddf(d) / L/mol\") ","category":"page"},{"location":"results/#Units","page":"Results","title":"Units","text":"","category":"section"},{"location":"results/","page":"Results","title":"Results","text":"The distance is assumed to be in Å, as this is the most common distance units in molecular simulations. The coordinates of the atoms are assumed be provided in Å. \nThe minimum-distance distribution function is unit-less, since it is the ratio of the density at each distance divided by an ideal-gas density.\nThe Kirkwood-Buff integrals are returned in cm³ mol⁻¹, if the coordinates were provided in Å.","category":"page"},{"location":"results/","page":"Results","title":"Results","text":"warning: Warning\nIf the coordinates are not in Å, the calculation will proceed normally, but the units of the KB integrals, which has units of volume per mol, should be converted to conform the length unit provided. ","category":"page"},{"location":"results/#Coordination-number-and-other-data","page":"Results","title":"Coordination number and other data","text":"","category":"section"},{"location":"results/","page":"Results","title":"Results","text":"Obtaining the MDDF involves the computation of some intermediate properties that are frequently useful for additional solution structure analysis. In particular, the coordination numbers are computed. For example, the coordination number as a function from the distance to the solute can be retrieved from a Results data structure with:","category":"page"},{"location":"results/","page":"Results","title":"Results","text":"coordination_number = results.coordination_number","category":"page"},{"location":"results/","page":"Results","title":"Results","text":"and this data can be plotted against the distances by:","category":"page"},{"location":"results/","page":"Results","title":"Results","text":"plot(result.d,results.coordination_number)","category":"page"},{"location":"results/","page":"Results","title":"Results","text":"The coordination number of subgroups can also be obtained, as explained in the Coordination number section.","category":"page"},{"location":"results/","page":"Results","title":"Results","text":"The complete data available is:","category":"page"},{"location":"results/","page":"Results","title":"Results","text":"Parameter Meaning Type of value Comment\nd Vector of distances of the histograms. Vector{Float64} To be used as the x coordinate on plotting any of the data.\nmd_count Non-normalized count of minimum distances at each d. Vector{Float64} This is the number of minimum distances found at each histogram bin, without normalization. Usually this is not interesting to analyze, because it is dependent on the bin size.\nmd_count_random Number of minimum distances found at each histogram bin for the random distribution. Vector{Float64} This is the normalization required to convert the md_count array into the minimum-distance distribution.\ncoordination_number Cumulative number of sites found for each histogram distance. Vector{Float64} This is the coordination number, that is, the number of sites found cumulative up to each distance, without any normalization.\ncoordination_number_random Cumulative site count for the random distribution. Vector{Float64} Usually not interesting for analysis.\nmddf The final distribution function. Vector{Float64} This is the MDDF computed (md_count normalized by md_count_random). It is the main result of the calculation.\nkb The final Kirkwood-Buff integral. Vector{Float64} This is the final KB integral, as a function of the integration distance from the solute. Computed as coordination_number - coordination_number_random\nsolute_atom Atomic contributions of the solute. Matrix{Float64} This is a matrix with nbins lines and solute.natomspermol columns, containing the atomic contributions of each solute atom to the complete MDDF.\nsolvent_atom Atomic contributions of the solvent. Matrix{Float64} This is a matrix with nbins lines and solvent.natomspermol columns, containing the atomic contributions of each solvent atom to the complete MDDF.\ndensity.solute Density (concentration) of the solute in the complete simulation box. Float64 In units of molecules/textrmAA^3\ndensity.solvent Density (concentration) of the solvent in the complete simulation box. Float64 In units of molecules/textrmAA^3\ndensity.solvent_bulk Density (concentration) of the solute in the bulk region. Float64 In units of molecules/textrmAA^3\nvolume Volume measures. Volume Contains the total volume of the simulation, the bulk volume, the volume of the solute domain and the shell volume of each bin of the histogram. These are computed by numerical integration from the random distributions.\nfiles List of files read. Vector{String} \nweights Weights of each file in the final counts. Vector{Float64} If the trajectories have different lengths or number of frames, the weights are adapted accordingly.\n ","category":"page"},{"location":"results/#Other-Result-parameters-available-which-are-set-at-Options:","page":"Results","title":"Other Result parameters available which are set at Options:","text":"","category":"section"},{"location":"results/","page":"Results","title":"Results","text":"Parameter Meaning Type of value Comment\nnbins Number of bins of the histograms. Int \ndbulk Distance from solute of bulk solution. Float64 \ncutoff Maximum distance to be considered for histograms. Float64 \nautocorrelation The solute is the same as the solvent? Bool Automatically set if solute == solvent.\nsolute Properties of the solute AtomSelection Contains the number of atoms, number of atoms per molecule and number of molecules of the solute.\nsolvent Properties of the solvent. AtomSelection Contains the number of atoms, number of atoms per molecule and number of molecules of the solvent.\nirefatom This is a reference atom that is used to generate random rotations and translations internally. Int Counts of the distributions for this atom are performed automatically to obtain radial (or proximal) distribution functions. Can be used for testing purposes.\nrdf_count This is the md_count minimum distance count of irefatom. Vector{Float64} This corresponds to the conventional radial distribution function if the solute contains only one atom.\nrdf_count_random Minimum distance of irefatom count for the random distribution. Vector{Float64} \nrdf Distribution function computed from the irefatom distribution. It is a conventional rdf if the solvent has only one atom. Vector{Float64} \nkb_rdf Kirkwood-Buff integral computed from the irefatom distribution. Vector{Float64} This must converge, at long distances, to the same value as kb, and can be used for testing.\noptions Calculation options. Options Carries (some redundant) options set by the user.\nlastframe_read Last frame read from the trajectory. Int \nn_frames_read Number of frames read from the trajectory. Int Can differ from lastframe_read if stride != 1\n ","category":"page"},{"location":"results/#Reference-functions","page":"Results","title":"Reference functions","text":"","category":"section"},{"location":"results/","page":"Results","title":"Results","text":"Modules = [ComplexMixtures]\nPages = [\"results.jl\"]","category":"page"},{"location":"results/#ComplexMixtures.Density","page":"Results","title":"ComplexMixtures.Density","text":"mutable struct Density\n\nStructure to contain the density values obtained from the calculation.\n\nsolute::Float64\nsolvent::Float64\nsolvent_bulk::Float64\n\n\n\n\n\n","category":"type"},{"location":"results/#ComplexMixtures.Result","page":"Results","title":"ComplexMixtures.Result","text":"mutable struct Result\n\nStructure to contain the results of the MDDF calculation.\n\nVersion::VersionNumber\nnbins::Int64\ndbulk::Float64\ncutoff::Float64\nd::Vector{Float64}\nmd_count::Vector{Float64}\nmd_count_random::Vector{Float64}\ncoordination_number::Vector{Float64}\ncoordination_number_random::Vector{Float64}\nmddf::Vector{Float64}\nkb::Vector{Float64}\nautocorrelation::Bool\nsolute::AtomSelection\nsolvent::AtomSelection\nsolute_group_count::Vector{Vector{Float64}}\nsolvent_group_count::Vector{Vector{Float64}}\nrdf_count::Vector{Float64}\nrdf_count_random::Vector{Float64}\nsum_rdf_count::Vector{Float64}\nsum_rdf_count_random::Vector{Float64}\nrdf::Vector{Float64}\nkb_rdf::Vector{Float64}\ndensity::ComplexMixtures.Density\nvolume::ComplexMixtures.Volume\nfiles::Vector{ComplexMixtures.TrajectoryFileOptions}\nweights::Vector{Float64}\n\nThe Result{Vector{Float64}} parametric type is necessary only for reading the JSON3 saved file. \n\n\n\n\n\n","category":"type"},{"location":"results/#Base.merge-Tuple{Vector{<:Result}}","page":"Results","title":"Base.merge","text":"merge(r::Vector{Result})\n\nThis function merges the results of MDDF calculations obtained by running the same analysis on multiple trajectories, or multiple parts of the same trajectory. It returns a Result structure of the same type, with all the functions and counters representing averages of the set provided weighted by the number of frames read in each Result set.\n\n\n\n\n\n","category":"method"},{"location":"results/#ComplexMixtures.load-Tuple{String}","page":"Results","title":"ComplexMixtures.load","text":"load(filename::String)\n\nFunction to load the json saved results file into the Result data structure.\n\n\n\n\n\n","category":"method"},{"location":"results/#ComplexMixtures.overview-Tuple{Result}","page":"Results","title":"ComplexMixtures.overview","text":"overview(R::Result)\n\nFunction that outputs the volumes and densities in the most natural units.\n\n\n\n\n\n","category":"method"},{"location":"results/#ComplexMixtures.save-Tuple{Result, String}","page":"Results","title":"ComplexMixtures.save","text":"save(R::Result, filename::String)\n\nFunction to write the result data structure to a json file.\n\n\n\n\n\n","category":"method"},{"location":"contributions/#contributions","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"","category":"section"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"One of the interesting features of Minimum-Distance distributions is that they can be naturally decomposed into the atomic or group contributions. Simply put, if a MDDF has a peak at a hydrogen-bonding distance, it is natural to decompose that peak into the contributions of each type of solute or solvent atom to that peak. ","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"To obtain the atomic contributions of an atom or group of atoms to the MDDF, the coordination number, or the site count at each distance, the contributions function is provided. For example, in a system composed of a protein and water, we would have defined the solute and solvent using:","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"using PDBTools, ComplexMixtures\natoms = readPDB(\"system.pdb\")\nprotein = select(atoms,\"protein\")\nwater = select(atoms,\"water\")\nsolute = AtomSelection(protein,nmols=1)\nsolvent = AtomSelection(water,natomspermol=3)","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"The MDDF calculation is executed with:","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"trajectory = Trajectory(\"trajectory.dcd\",solute,solvent)\nresults = mddf(trajectory, Options(bulk_range=(8.0, 12.0)))","category":"page"},{"location":"contributions/#Atomic-contributions-in-the-result-data-structure","page":"Atomic and group contributions","title":"Atomic contributions in the result data structure","text":"","category":"section"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"The results data structure contains the decomposition of the MDDF into the contributions of every type of atom of the solute and the solvent. These contributions can be retrieved using the contributions function, with the SoluteGroup and SolventGroup selectors.","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"For example, if the MDDF of water (solvent) relative to a solute was computed, and water has atom names OH2, H1, H2, one can retrieve the contributions of the oxygen atom with:","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"OH2 = contributions(results, SolventGroup([\"OH2\"]))","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"or with, if OH2 is the first atom in the molecule,","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"OH2 = contributions(results, SolventGroup([1]))","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"The contributions of the hydrogen atoms can be obtained, similarly, with:","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"H = contributions(results, SolventGroup([\"H1\", \"H2\"]))","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"or with, if OH2 is the first atom in the molecule,","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"H = contributions(results, SolventGroup([2, 3]))","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"Each of these calls will return a vector of the constributions of these atoms to the total MDDF. ","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"For example, here we plot the total MDDF and the Oxygen contributions: ","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"using Plots\nplot(results.d, results.mddf, label=[\"Total MDDF\"], linewidth=2)\nplot!(results.d, contributions(results, SolventGroup([\"OH2\"])), label=[\"OH2\"], linewidth=2)\nplot!(xlabel=\"Distance / Å\", ylabel=\"MDDF\")","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"
","category":"page"},{"location":"results/","page":"Results","title":"Results","text":"Thus, this plot was obtained with the following code:","category":"page"},{"location":"results/","page":"Results","title":"Results","text":"using Plots\nplot(results.d,results.kb,xlabel=\"d/A\",ylabel=\"mddf(d) / L/mol\") ","category":"page"},{"location":"results/#Units","page":"Results","title":"Units","text":"","category":"section"},{"location":"results/","page":"Results","title":"Results","text":"The distance is assumed to be in Å, as this is the most common distance units in molecular simulations. The coordinates of the atoms are assumed be provided in Å. \nThe minimum-distance distribution function is unit-less, since it is the ratio of the density at each distance divided by an ideal-gas density.\nThe Kirkwood-Buff integrals are returned in cm³ mol⁻¹, if the coordinates were provided in Å.","category":"page"},{"location":"results/","page":"Results","title":"Results","text":"warning: Warning\nIf the coordinates are not in Å, the calculation will proceed normally, but the units of the KB integrals, which has units of volume per mol, should be converted to conform the length unit provided. ","category":"page"},{"location":"results/#Coordination-number-and-other-data","page":"Results","title":"Coordination number and other data","text":"","category":"section"},{"location":"results/","page":"Results","title":"Results","text":"Obtaining the MDDF involves the computation of some intermediate properties that are frequently useful for additional solution structure analysis. In particular, the coordination numbers are computed. For example, the coordination number as a function from the distance to the solute can be retrieved from a Results data structure with:","category":"page"},{"location":"results/","page":"Results","title":"Results","text":"coordination_number = results.coordination_number","category":"page"},{"location":"results/","page":"Results","title":"Results","text":"and this data can be plotted against the distances by:","category":"page"},{"location":"results/","page":"Results","title":"Results","text":"plot(result.d,results.coordination_number)","category":"page"},{"location":"results/","page":"Results","title":"Results","text":"The coordination number of subgroups can also be obtained, as explained in the Coordination number section.","category":"page"},{"location":"results/","page":"Results","title":"Results","text":"The complete data available is:","category":"page"},{"location":"results/","page":"Results","title":"Results","text":"Parameter Meaning Type of value Comment\nd Vector of distances of the histograms. Vector{Float64} To be used as the x coordinate on plotting any of the data.\nmd_count Non-normalized count of minimum distances at each d. Vector{Float64} This is the number of minimum distances found at each histogram bin, without normalization. Usually this is not interesting to analyze, because it is dependent on the bin size.\nmd_count_random Number of minimum distances found at each histogram bin for the random distribution. Vector{Float64} This is the normalization required to convert the md_count array into the minimum-distance distribution.\ncoordination_number Cumulative number of sites found for each histogram distance. Vector{Float64} This is the coordination number, that is, the number of sites found cumulative up to each distance, without any normalization.\ncoordination_number_random Cumulative site count for the random distribution. Vector{Float64} Usually not interesting for analysis.\nmddf The final distribution function. Vector{Float64} This is the MDDF computed (md_count normalized by md_count_random). It is the main result of the calculation.\nkb The final Kirkwood-Buff integral. Vector{Float64} This is the final KB integral, as a function of the integration distance from the solute. Computed as coordination_number - coordination_number_random\nsolute_atom Atomic contributions of the solute. Matrix{Float64} This is a matrix with nbins lines and solute.natomspermol columns, containing the atomic contributions of each solute atom to the complete MDDF.\nsolvent_atom Atomic contributions of the solvent. Matrix{Float64} This is a matrix with nbins lines and solvent.natomspermol columns, containing the atomic contributions of each solvent atom to the complete MDDF.\ndensity.solute Density (concentration) of the solute in the complete simulation box. Float64 In units of molecules/textrmAA^3\ndensity.solvent Density (concentration) of the solvent in the complete simulation box. Float64 In units of molecules/textrmAA^3\ndensity.solvent_bulk Density (concentration) of the solute in the bulk region. Float64 In units of molecules/textrmAA^3\nvolume Volume measures. Volume Contains the total volume of the simulation, the bulk volume, the volume of the solute domain and the shell volume of each bin of the histogram. These are computed by numerical integration from the random distributions.\nfiles List of files read. Vector{String} \nweights Weights of each file in the final counts. Vector{Float64} If the trajectories have different lengths or number of frames, the weights are adapted accordingly.\n ","category":"page"},{"location":"results/#Other-Result-parameters-available-which-are-set-at-Options:","page":"Results","title":"Other Result parameters available which are set at Options:","text":"","category":"section"},{"location":"results/","page":"Results","title":"Results","text":"Parameter Meaning Type of value Comment\nnbins Number of bins of the histograms. Int \ndbulk Distance from solute of bulk solution. Float64 \ncutoff Maximum distance to be considered for histograms. Float64 \nautocorrelation The solute is the same as the solvent? Bool Automatically set if solute == solvent.\nsolute Properties of the solute AtomSelection Contains the number of atoms, number of atoms per molecule and number of molecules of the solute.\nsolvent Properties of the solvent. AtomSelection Contains the number of atoms, number of atoms per molecule and number of molecules of the solvent.\nirefatom This is a reference atom that is used to generate random rotations and translations internally. Int Counts of the distributions for this atom are performed automatically to obtain radial (or proximal) distribution functions. Can be used for testing purposes.\nrdf_count This is the md_count minimum distance count of irefatom. Vector{Float64} This corresponds to the conventional radial distribution function if the solute contains only one atom.\nrdf_count_random Minimum distance of irefatom count for the random distribution. Vector{Float64} \nrdf Distribution function computed from the irefatom distribution. It is a conventional rdf if the solvent has only one atom. Vector{Float64} \nkb_rdf Kirkwood-Buff integral computed from the irefatom distribution. Vector{Float64} This must converge, at long distances, to the same value as kb, and can be used for testing.\noptions Calculation options. Options Carries (some redundant) options set by the user.\nlastframe_read Last frame read from the trajectory. Int \nn_frames_read Number of frames read from the trajectory. Int Can differ from lastframe_read if stride != 1\n ","category":"page"},{"location":"results/#Reference-functions","page":"Results","title":"Reference functions","text":"","category":"section"},{"location":"results/","page":"Results","title":"Results","text":"Modules = [ComplexMixtures]\nPages = [\"results.jl\"]","category":"page"},{"location":"results/#ComplexMixtures.Density","page":"Results","title":"ComplexMixtures.Density","text":"mutable struct Density\n\nStructure to contain the density values obtained from the calculation.\n\nsolute::Float64\nsolvent::Float64\nsolvent_bulk::Float64\n\n\n\n\n\n","category":"type"},{"location":"results/#ComplexMixtures.Result","page":"Results","title":"ComplexMixtures.Result","text":"mutable struct Result\n\nStructure to contain the results of the MDDF calculation.\n\nVersion::VersionNumber\nnbins::Int64\ndbulk::Float64\ncutoff::Float64\nd::Vector{Float64}\nmd_count::Vector{Float64}\nmd_count_random::Vector{Float64}\ncoordination_number::Vector{Float64}\ncoordination_number_random::Vector{Float64}\nmddf::Vector{Float64}\nkb::Vector{Float64}\nautocorrelation::Bool\nsolute::AtomSelection\nsolvent::AtomSelection\nsolute_group_count::Vector{Vector{Float64}}\nsolvent_group_count::Vector{Vector{Float64}}\nrdf_count::Vector{Float64}\nrdf_count_random::Vector{Float64}\nsum_rdf_count::Vector{Float64}\nsum_rdf_count_random::Vector{Float64}\nrdf::Vector{Float64}\nkb_rdf::Vector{Float64}\ndensity::ComplexMixtures.Density\nvolume::ComplexMixtures.Volume\nfiles::Vector{ComplexMixtures.TrajectoryFileOptions}\nweights::Vector{Float64}\n\nThe Result{Vector{Float64}} parametric type is necessary only for reading the JSON3 saved file. \n\n\n\n\n\n","category":"type"},{"location":"results/#Base.merge-Tuple{Vector{<:Result}}","page":"Results","title":"Base.merge","text":"merge(r::Vector{Result})\n\nThis function merges the results of MDDF calculations obtained by running the same analysis on multiple trajectories, or multiple parts of the same trajectory. It returns a Result structure of the same type, with all the functions and counters representing averages of the set provided weighted by the number of frames read in each Result set.\n\n\n\n\n\n","category":"method"},{"location":"results/#ComplexMixtures.load-Tuple{String}","page":"Results","title":"ComplexMixtures.load","text":"load(filename::String)\n\nFunction to load the json saved results file into the Result data structure.\n\n\n\n\n\n","category":"method"},{"location":"results/#ComplexMixtures.overview-Tuple{Result}","page":"Results","title":"ComplexMixtures.overview","text":"overview(R::Result)\n\nFunction that outputs the volumes and densities in the most natural units.\n\n\n\n\n\n","category":"method"},{"location":"results/#ComplexMixtures.save-Tuple{Result, String}","page":"Results","title":"ComplexMixtures.save","text":"save(R::Result, filename::String)\n\nFunction to write the result data structure to a json file.\n\n\n\n\n\n","category":"method"},{"location":"contributions/#contributions","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"","category":"section"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"One of the interesting features of Minimum-Distance distributions is that they can be naturally decomposed into the atomic or group contributions. Simply put, if a MDDF has a peak at a hydrogen-bonding distance, it is natural to decompose that peak into the contributions of each type of solute or solvent atom to that peak. ","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"To obtain the atomic contributions of an atom or group of atoms to the MDDF, the coordination number, or the site count at each distance, the contributions function is provided. For example, in a system composed of a protein and water, we would have defined the solute and solvent using:","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"using PDBTools, ComplexMixtures\natoms = readPDB(\"system.pdb\")\nprotein = select(atoms,\"protein\")\nwater = select(atoms,\"water\")\nsolute = AtomSelection(protein,nmols=1)\nsolvent = AtomSelection(water,natomspermol=3)","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"The MDDF calculation is executed with:","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"trajectory = Trajectory(\"trajectory.dcd\",solute,solvent)\nresults = mddf(trajectory, Options(bulk_range=(8.0, 12.0)))","category":"page"},{"location":"contributions/#Atomic-contributions-in-the-result-data-structure","page":"Atomic and group contributions","title":"Atomic contributions in the result data structure","text":"","category":"section"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"The results data structure contains the decomposition of the MDDF into the contributions of every type of atom of the solute and the solvent. These contributions can be retrieved using the contributions function, with the SoluteGroup and SolventGroup selectors.","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"For example, if the MDDF of water (solvent) relative to a solute was computed, and water has atom names OH2, H1, H2, one can retrieve the contributions of the oxygen atom with:","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"OH2 = contributions(results, SolventGroup([\"OH2\"]))","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"or with, if OH2 is the first atom in the molecule,","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"OH2 = contributions(results, SolventGroup([1]))","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"The contributions of the hydrogen atoms can be obtained, similarly, with:","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"H = contributions(results, SolventGroup([\"H1\", \"H2\"]))","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"or with, if OH2 is the first atom in the molecule,","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"H = contributions(results, SolventGroup([2, 3]))","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"Each of these calls will return a vector of the constributions of these atoms to the total MDDF. ","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"For example, here we plot the total MDDF and the Oxygen contributions: ","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"using Plots\nplot(results.d, results.mddf, label=[\"Total MDDF\"], linewidth=2)\nplot!(results.d, contributions(results, SolventGroup([\"OH2\"])), label=[\"OH2\"], linewidth=2)\nplot!(xlabel=\"Distance / Å\", ylabel=\"MDDF\")","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":" ","category":"page"},{"location":"contributions/#Contributions-to-coordination-numbers-or-site-counts","page":"Atomic and group contributions","title":"Contributions to coordination numbers or site counts","text":"","category":"section"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"The keyword type defines the return type of the contribution:","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"type=:mddf : the contribution of the group to the MDDF is returned (default).\ntype=:coordination_number : the contribution of the group to the coordination number, that is, the cumulative sum of counts at each distance, is returned.\ntype=:md_count : the contribution of the group to the site count at each distance is returned. ","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"Example of the usage of the type option:","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"ca_contributions = contributions(results, SoluteGroup([\"CA\"]); type=:coordination_number)","category":"page"},{"location":"contributions/#Using-PDBTools","page":"Atomic and group contributions","title":"Using PDBTools","text":"","category":"section"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"If the solute is a protein, or other complex molecule, selections defined with PDBTools can be used. For example, this will retrieve the contribution of the acidic residues of a protein to total MDDF:","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"using PDBTools\natoms = readPDB(\"system.pdb\")\nacidic_residues = select(atoms, \"acidic\")\nacidic_contributions = contributions(results, SoluteGroup(acidic_residues))","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"It is expected that for a protein most of the atoms do not contribute to the MDDF, and that all values are zero at very short distances, smaller than the radii of the atoms.","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"More interesting and general is to select atoms of a complex molecule, like a protein, using residue names, types, etc. Here we illustrate how this is done by providing selection strings to contributions to obtain the contributions to the MDDF of different types of residues of a protein to the total MDDF. ","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"For example, if we want to split the contributions of the charged and neutral residues to the total MDDF distribution, we could use to following code. Here, solute refers to the protein.","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"charged_residues = PDBTools.select(atoms,\"charged\")\ncharged_contributions = contributions(results, SoluteGroup(charged_residues))\n\nneutral_residues = PDBTools.select(atoms,\"neutral\")\nneutral_contributions = contributions(atoms, SoluteGroup(neutral_residues))","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"The charged_contributions and neutral_contributions outputs are vectors containing the contributions of these residues to the total MDDF. The corresponding plot is: ","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"plot(results.d,results.mddf,label=\"Total MDDF\",linewidth=2)\nplot!(results.d,charged_contributions,label=\"Charged residues\",linewidth=2)\nplot!(results.d,neutral_contributions,label=\"Neutral residues\",linewidth=2)\nplot!(xlabel=\"Distance / Å\",ylabel=\"MDDF\")","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"Resulting in:","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"
","category":"page"},{"location":"contributions/#Contributions-to-coordination-numbers-or-site-counts","page":"Atomic and group contributions","title":"Contributions to coordination numbers or site counts","text":"","category":"section"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"The keyword type defines the return type of the contribution:","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"type=:mddf : the contribution of the group to the MDDF is returned (default).\ntype=:coordination_number : the contribution of the group to the coordination number, that is, the cumulative sum of counts at each distance, is returned.\ntype=:md_count : the contribution of the group to the site count at each distance is returned. ","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"Example of the usage of the type option:","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"ca_contributions = contributions(results, SoluteGroup([\"CA\"]); type=:coordination_number)","category":"page"},{"location":"contributions/#Using-PDBTools","page":"Atomic and group contributions","title":"Using PDBTools","text":"","category":"section"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"If the solute is a protein, or other complex molecule, selections defined with PDBTools can be used. For example, this will retrieve the contribution of the acidic residues of a protein to total MDDF:","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"using PDBTools\natoms = readPDB(\"system.pdb\")\nacidic_residues = select(atoms, \"acidic\")\nacidic_contributions = contributions(results, SoluteGroup(acidic_residues))","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"It is expected that for a protein most of the atoms do not contribute to the MDDF, and that all values are zero at very short distances, smaller than the radii of the atoms.","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"More interesting and general is to select atoms of a complex molecule, like a protein, using residue names, types, etc. Here we illustrate how this is done by providing selection strings to contributions to obtain the contributions to the MDDF of different types of residues of a protein to the total MDDF. ","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"For example, if we want to split the contributions of the charged and neutral residues to the total MDDF distribution, we could use to following code. Here, solute refers to the protein.","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"charged_residues = PDBTools.select(atoms,\"charged\")\ncharged_contributions = contributions(results, SoluteGroup(charged_residues))\n\nneutral_residues = PDBTools.select(atoms,\"neutral\")\nneutral_contributions = contributions(atoms, SoluteGroup(neutral_residues))","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"The charged_contributions and neutral_contributions outputs are vectors containing the contributions of these residues to the total MDDF. The corresponding plot is: ","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"plot(results.d,results.mddf,label=\"Total MDDF\",linewidth=2)\nplot!(results.d,charged_contributions,label=\"Charged residues\",linewidth=2)\nplot!(results.d,neutral_contributions,label=\"Neutral residues\",linewidth=2)\nplot!(xlabel=\"Distance / Å\",ylabel=\"MDDF\")","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"Resulting in:","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":" ","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"Note here how charged residues contribute strongly to the peak at hydrogen-bonding distances, but much less in general. Of course all selection options could be used, to obtain the contributions of specific types of residues, atoms, the backbone, the side-chains, etc. ","category":"page"},{"location":"contributions/#Reference-functions","page":"Atomic and group contributions","title":"Reference functions","text":"","category":"section"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"Modules = [ComplexMixtures]\nPages = [\"contributions.jl\"]","category":"page"},{"location":"contributions/#ComplexMixtures.contributions-Tuple{Result, Union{SoluteGroup, SolventGroup}}","page":"Atomic and group contributions","title":"ComplexMixtures.contributions","text":"contributions(R::Result, group::Union{SoluteGroup,SolventGroup}; type = :mddf)\n\nReturns the contributions of the atoms of the solute or solvent to the MDDF, coordination number, or MD count.\n\nArguments\n\nR::Result: The result of a calculation.\ngroup::Union{SoluteGroup,SolventGroup}: The group of atoms to consider.\ntype::Symbol: The type of contributions to return. Can be :mddf (default), :coordination_number, or :md_count.\n\nExamples\n\njulia> using ComplexMixtures, PDBTools\n\njulia> dir = ComplexMixtures.Testing.data_dir*\"/Gromacs\";\n\njulia> atoms = readPDB(dir*\"/system.pdb\");\n\njulia> protein = select(atoms, \"protein\");\n\njulia> emim = select(atoms, \"resname EMI\"); \n\njulia> solute = AtomSelection(protein, nmols = 1)\nAtomSelection \n 1231 atoms belonging to 1 molecule(s).\n Atoms per molecule: 1231\n Number of groups: 1231\n\njulia> solvent = AtomSelection(emim, natomspermol = 20)\nAtomSelection \n 5080 atoms belonging to 254 molecule(s).\n Atoms per molecule: 20\n Number of groups: 20\n\njulia> results = load(dir*\"/protein_EMI.json\"); # load pre-calculated results\n\njulia> ca_cb = contributions(results, SoluteGroup([\"CA\", \"CB\"])); # contribution of CA and CB atoms to the MDDF\n\njulia> ca_cb = contributions(results, SoluteGroup([\"CA\", \"CB\"]); type=:coordination_number); # contribution of CA and CB atoms to the coordination number\n\n\n\n\n\n","category":"method"},{"location":"example1/#Protein-in-water/glycerol","page":"◦ Protein in water/glycerol","title":"Protein in water/glycerol","text":"","category":"section"},{"location":"example1/","page":"◦ Protein in water/glycerol","title":"◦ Protein in water/glycerol","text":"The following examples consider a system composed a protein solvated by a mixture of water and glycerol, built with Packmol. The simulations were performed with NAMD with periodic boundary conditions and a NPT ensemble at room temperature and pressure. Molecular pictures were produced with VMD and plots were produced with Julia's Plots library.","category":"page"},{"location":"example1/","page":"◦ Protein in water/glycerol","title":"◦ Protein in water/glycerol","text":"
","category":"page"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"Note here how charged residues contribute strongly to the peak at hydrogen-bonding distances, but much less in general. Of course all selection options could be used, to obtain the contributions of specific types of residues, atoms, the backbone, the side-chains, etc. ","category":"page"},{"location":"contributions/#Reference-functions","page":"Atomic and group contributions","title":"Reference functions","text":"","category":"section"},{"location":"contributions/","page":"Atomic and group contributions","title":"Atomic and group contributions","text":"Modules = [ComplexMixtures]\nPages = [\"contributions.jl\"]","category":"page"},{"location":"contributions/#ComplexMixtures.contributions-Tuple{Result, Union{SoluteGroup, SolventGroup}}","page":"Atomic and group contributions","title":"ComplexMixtures.contributions","text":"contributions(R::Result, group::Union{SoluteGroup,SolventGroup}; type = :mddf)\n\nReturns the contributions of the atoms of the solute or solvent to the MDDF, coordination number, or MD count.\n\nArguments\n\nR::Result: The result of a calculation.\ngroup::Union{SoluteGroup,SolventGroup}: The group of atoms to consider.\ntype::Symbol: The type of contributions to return. Can be :mddf (default), :coordination_number, or :md_count.\n\nExamples\n\njulia> using ComplexMixtures, PDBTools\n\njulia> dir = ComplexMixtures.Testing.data_dir*\"/Gromacs\";\n\njulia> atoms = readPDB(dir*\"/system.pdb\");\n\njulia> protein = select(atoms, \"protein\");\n\njulia> emim = select(atoms, \"resname EMI\"); \n\njulia> solute = AtomSelection(protein, nmols = 1)\nAtomSelection \n 1231 atoms belonging to 1 molecule(s).\n Atoms per molecule: 1231\n Number of groups: 1231\n\njulia> solvent = AtomSelection(emim, natomspermol = 20)\nAtomSelection \n 5080 atoms belonging to 254 molecule(s).\n Atoms per molecule: 20\n Number of groups: 20\n\njulia> results = load(dir*\"/protein_EMI.json\"); # load pre-calculated results\n\njulia> ca_cb = contributions(results, SoluteGroup([\"CA\", \"CB\"])); # contribution of CA and CB atoms to the MDDF\n\njulia> ca_cb = contributions(results, SoluteGroup([\"CA\", \"CB\"]); type=:coordination_number); # contribution of CA and CB atoms to the coordination number\n\n\n\n\n\n","category":"method"},{"location":"example1/#Protein-in-water/glycerol","page":"◦ Protein in water/glycerol","title":"Protein in water/glycerol","text":"","category":"section"},{"location":"example1/","page":"◦ Protein in water/glycerol","title":"◦ Protein in water/glycerol","text":"The following examples consider a system composed a protein solvated by a mixture of water and glycerol, built with Packmol. The simulations were performed with NAMD with periodic boundary conditions and a NPT ensemble at room temperature and pressure. Molecular pictures were produced with VMD and plots were produced with Julia's Plots library.","category":"page"},{"location":"example1/","page":"◦ Protein in water/glycerol","title":"◦ Protein in water/glycerol","text":" \n
\n \n
\n \n
\n \n
\n \n
\n \n
\n \n
\n \n
\n

 \n
\n \n
\n \n
\n \n
\n \n
\n \n
\n \n
\n \n
\n \n
\n +
+ +
+ +
+