+ {% for id in include.contributors.reviewing %}{{ include.sep }}{% if include.badge %}{% include _includes/contributor-badge.html id=id small=true %}{% else %}{{ contributors[id].name | default: id }}{% endif %}{%- endfor -%}
+
+
+ {% endif %}
+
You can view the tutorial materials in different languages by clicking the dropdown icon next to the slides ({% icon slides %}) and tutorial ({% icon tutorial %}) buttons below.
{% if topic.draft %}
diff --git a/_layouts/workflow-list.html b/_layouts/workflow-list.html
index b5a1e5732fa54..4d0be04cfedc2 100644
--- a/_layouts/workflow-list.html
+++ b/_layouts/workflow-list.html
@@ -4,71 +4,44 @@
{% assign material = site | fetch_tutorial_material:page.topic_name,page.tutorial_name%}
-
-
To use these workflows in Galaxy you can either click the links to download the workflows, or you can right-click and copy the link to the workflow which can be used in the Galaxy form to import workflows.
-
-
-
- {% for workflow in material.workflows %}
-
-
-
-
{{ workflow.title }}
-
- {% for entity in workflow.creators %}
- {{ entity.name }}{% if forloop.last == false %}, {% endif %}
- {% endfor %}
-
- Below are the instructions for importing these workflows directly into your Galaxy server of choice to start using them!
-
- {% snippet faqs/galaxy/workflows_import.md box_type="hands_on" %}
-
+
diff --git a/_plugins/author-page.rb b/_plugins/author-page.rb
index 2f002c142e880..a82bccdfcff80 100644
--- a/_plugins/author-page.rb
+++ b/_plugins/author-page.rb
@@ -1,5 +1,6 @@
# frozen_string_literal: true
+require './_plugins/gtn/mod'
require './_plugins/gtn'
module Jekyll
@@ -168,3 +169,70 @@ def generate(site)
end
end
end
+
+Jekyll::Hooks.register :site, :post_read do |site|
+ if Jekyll.env == 'production'
+ Jekyll.logger.info "[GTN/Reviewers] Ingesting GitHub reviewer metadata"
+ start_time = Time.now
+ # Maps a lowercase version of their name to the potential mixed-case version
+ contribs_lower = site.data['contributors'].map{|k, v| [k.downcase, k]}.to_h
+
+ # Annotate the from github metadata
+ gh_reviewers_by_path = Hash.new { |hash, key| hash[key] = [] }
+ # Hash of PRs by path
+ site.data['github'].each do |num, pr|
+ # Within a PR we have some reviews, let's get that set organised:
+ reviewers = pr['reviews'].map do |review|
+ # Just "people"
+ contribs_lower.fetch(review['author']['login'].downcase, review['author']['login'])
+ end.uniq.reject{|x| x == 'github-actions'}
+
+ pr['files'].select{|p| p['path'] =~ /.(md|html)$/}.each do |file|
+ real_path = Gtn::PublicationTimes.chase_rename(file['path'])
+ gh_reviewers_by_path[real_path] += reviewers
+ gh_reviewers_by_path[real_path].uniq!
+ end
+ end
+
+ # For all of our pages, if the path is mentioned above, then, tag it.
+ site.pages.select{|t| gh_reviewers_by_path.key?(t.path)}.each do |t|
+ if t['layout'] == 'tutorial_hands_on' or !%w[base_slides introduction_slides tutorial_slides].index(t['layout']).nil?
+ if t.data.key?('contributors')
+ # Automatically 'upgrade' to new structure
+ t.data['contributions'] = {
+ 'authorship' => t.data['contributors'],
+ 'reviewing' => gh_reviewers_by_path[t.path]
+ }
+ t.data.delete('contributors')
+ elsif t.data.key?('contributions')
+ if t.data['contributions'].key?('reviewing')
+ t.data['contributions']['reviewing'] += gh_reviewers_by_path[t.path]
+ else
+ t.data['contributions']['reviewing'] = gh_reviewers_by_path[t.path]
+ end
+ t.data['contributions']['reviewing'].uniq!
+ end
+ end
+ end
+
+ site.posts.docs.select{|t| gh_reviewers_by_path.key?(t.path)}.each do |t|
+ if t['layout'] == 'news'
+ if t.data.key?('contributors')
+ t.data['contributions'] = {
+ 'authorship' => t.data['contributors'],
+ 'reviewing' => gh_reviewers_by_path[t.path]
+ }
+ t.data.delete('contributors')
+ elsif t.data.key?('contributions')
+ if t.data['contributions'].key?('reviewing')
+ t.data['contributions']['reviewing'] += gh_reviewers_by_path[t.path]
+ else
+ t.data['contributions']['reviewing'] = gh_reviewers_by_path[t.path]
+ end
+ t.data['contributions']['reviewing'].uniq!
+ end
+ end
+ end
+ Jekyll.logger.info "[GTN/Reviewers] Complete in #{Time.now - start_time} seconds"
+ end
+end
diff --git a/_plugins/gtn/contributors.rb b/_plugins/gtn/contributors.rb
index 56da53fbea3de..349349aae1855 100644
--- a/_plugins/gtn/contributors.rb
+++ b/_plugins/gtn/contributors.rb
@@ -6,6 +6,8 @@
module Gtn
# Parse the git repo to get some facts
module Contributors
+ @HAS_WARNED_ON = []
+
##
# Returns contributors, regardless of whether they are 'contributor' or 'contributions' style
# Params:
@@ -150,7 +152,10 @@ def self.fetch(site, c, warn: false)
return ['grant', site.data['grants'][c]]
else
if ! warn
- Jekyll.logger.warn "Contributor #{c} not found"
+ if ! @HAS_WARNED_ON.include?(c)
+ Jekyll.logger.warn "Contributor #{c} not found"
+ @HAS_WARNED_ON.push(c)
+ end
end
end
diff --git a/_plugins/gtn/mod.rb b/_plugins/gtn/mod.rb
index cbaeca0e34eec..f9c9af71b9319 100644
--- a/_plugins/gtn/mod.rb
+++ b/_plugins/gtn/mod.rb
@@ -115,9 +115,14 @@ def self.obtain_time(f_unk)
# This is faster than talking to the file system.
module PublicationTimes
@@TIME_CACHE = nil
+ @@RENAMES = nil
- def self.chase_rename(renames, path, depth: 0)
- if renames.key? path
+ def self.chase_rename(path, depth: 0)
+ if @@RENAMES.nil?
+ self.init_cache
+ end
+
+ if @@RENAMES.key? path
# TODO(hexylena)
# This happens because it's the wrong datastructure, if there's a loop
# in there, it'll just cycle through it endlessly.
@@ -128,7 +133,7 @@ def self.chase_rename(renames, path, depth: 0)
Jekyll.logger.error "[GTN/Time/Pub] Too many renames for #{path}"
path
else
- chase_rename(renames, renames[path], depth: depth + 1)
+ chase_rename(@@RENAMES[path], depth: depth + 1)
end
else
path
@@ -139,7 +144,7 @@ def self.init_cache
return unless @@TIME_CACHE.nil?
@@TIME_CACHE = {}
- renames = {}
+ @@RENAMES = {}
Jekyll.logger.info '[GTN/Time/Pub] Filling Publication Time Cache'
cached_command
@@ -151,11 +156,11 @@ def self.init_cache
modification_type, path = f.split("\t")
if modification_type == 'A'
# Chase the renames.
- final_filename = chase_rename(renames, path)
+ final_filename = chase_rename(path)
@@TIME_CACHE[final_filename] = Time.at(date.to_i)
elsif modification_type[0] == 'R'
_, moved_from, moved_to = f.split("\t")
- renames[moved_from] = moved_to # Point from the 'older' version to the newer.
+ @@RENAMES[moved_from] = moved_to # Point from the 'older' version to the newer.
end
end
end
diff --git a/assets/css/main.scss b/assets/css/main.scss
index b338788a4710b..7be7c37fd12a9 100644
--- a/assets/css/main.scss
+++ b/assets/css/main.scss
@@ -1180,6 +1180,17 @@ nav[data-toggle='toc'] {
}
}
+ .reviewers {
+ .contributor-badge {
+ margin: 0;
+ padding: 0;
+ img {
+ margin: 0;
+ padding: 0;
+ }
+ }
+ }
+
}
.contributor-badge {
diff --git a/assets/css/slides.scss b/assets/css/slides.scss
index ca4625853a463..b32c34f798693 100644
--- a/assets/css/slides.scss
+++ b/assets/css/slides.scss
@@ -83,47 +83,47 @@ body.remark-container {
}
.image-05 img {
- width: 5%;
+ min-width: 5%;
}
.image-10 img {
- width: 10%;
+ min-width: 10%;
}
.image-15 img {
- width: 15%;
+ min-width: 15%;
}
.image-25 img {
- width: 25%;
+ min-width: 25%;
}
.image-40 img {
- width: 40%;
+ min-width: 40%;
}
.image-45 img {
- width: 45%;
+ min-width: 45%;
}
.image-50 img {
- width: 50%;
+ min-width: 50%;
}
.image-60 img {
- width: 60%;
+ min-width: 60%;
}
.image-75 img {
- width: 75%;
+ min-width: 75%;
}
.image-90 img {
- width: 100%;
+ min-width: 100%;
}
.image-100 img {
- width: 100%;
+ min-width: 100%;
}
.table-100 table {
@@ -360,7 +360,7 @@ body.remark-container {
.pull-right-small {
float: right;
width: 33%;
-
+
}
.pull-left {
diff --git a/assets/images/galaxy_climate.png b/assets/images/galaxy_climate.png
new file mode 100644
index 0000000000000..b993f714fcd5e
Binary files /dev/null and b/assets/images/galaxy_climate.png differ
diff --git a/assets/images/galaxy_subdomain.png b/assets/images/galaxy_subdomain.png
new file mode 100644
index 0000000000000..67d7d1f985735
Binary files /dev/null and b/assets/images/galaxy_subdomain.png differ
diff --git a/bin/check-url-persistence.sh b/bin/check-url-persistence.sh
index f9c7372cbc2b8..96be6e3436b97 100755
--- a/bin/check-url-persistence.sh
+++ b/bin/check-url-persistence.sh
@@ -24,7 +24,7 @@ cat /tmp/20*.txt | sort -u | \
grep --extended-regexp -v 'krona_?[a-z]*.html' | \
grep -v '/transcriptomics/tutorials/ref-based/faqs/rnaseq_data.html' | \
grep -v '/topics/data-management/' | \
- grep -v 'training-material/tags/' | grep -v 'data-library'| \
+ grep -v 'training-material/tags/' | grep -v 'data-library'| grep -v '/recordings/index.html' |\
sed 's|/$|/index.html|' | grep '.html$' | sort -u | sed 's|https://training.galaxyproject.org|_site|' > /tmp/gtn-files.txt
count=0
diff --git a/bin/schema-event-external.yaml b/bin/schema-event-external.yaml
index 973c5b02d6e0a..e27aa67106d8d 100644
--- a/bin/schema-event-external.yaml
+++ b/bin/schema-event-external.yaml
@@ -59,6 +59,13 @@ mapping:
- CONTRIBUTORS
- ORGANISATIONS
- GRANTS
+ reviewing:
+ type: seq
+ description: This person reviewed this material for accuracy and correctness
+ sequence:
+ - type: str
+ enum:
+ - CONTRIBUTORS
funding:
type: seq
description: These entities provided funding support for the development of this resource
diff --git a/bin/schema-event.yaml b/bin/schema-event.yaml
index 98d3437a5e207..52c6f69ca1336 100644
--- a/bin/schema-event.yaml
+++ b/bin/schema-event.yaml
@@ -54,6 +54,13 @@ mapping:
- CONTRIBUTORS
- ORGANISATIONS
- GRANTS
+ reviewing:
+ type: seq
+ description: This person reviewed this material for accuracy and correctness
+ sequence:
+ - type: str
+ enum:
+ - CONTRIBUTORS
funding:
type: seq
description: These entities provided funding support for the development of this resource
diff --git a/bin/schema-learning-pathway.yaml b/bin/schema-learning-pathway.yaml
index 018cbdd2596ad..5c18932b46f7d 100644
--- a/bin/schema-learning-pathway.yaml
+++ b/bin/schema-learning-pathway.yaml
@@ -123,6 +123,7 @@ mapping:
- ai4life
- assembly
- climate
+ - community
- computational-chemistry
- contributing
- data-science
diff --git a/bin/schema-news.yaml b/bin/schema-news.yaml
index e7ed0c10945eb..f1cd754c765fe 100644
--- a/bin/schema-news.yaml
+++ b/bin/schema-news.yaml
@@ -105,6 +105,13 @@ mapping:
- CONTRIBUTORS
- ORGANISATIONS
- GRANTS
+ reviewing:
+ type: seq
+ description: This person reviewed this material for accuracy and correctness
+ sequence:
+ - type: str
+ enum:
+ - CONTRIBUTORS
funding:
type: seq
description: These entities provided funding support for the development of this resource
diff --git a/bin/schema-slides.yaml b/bin/schema-slides.yaml
index d9859831a49e3..875579e19e019 100644
--- a/bin/schema-slides.yaml
+++ b/bin/schema-slides.yaml
@@ -154,6 +154,13 @@ mapping:
- CONTRIBUTORS
- ORGANISATIONS
- GRANTS
+ reviewing:
+ type: seq
+ description: This person reviewed this material for accuracy and correctness
+ sequence:
+ - type: str
+ enum:
+ - CONTRIBUTORS
funding:
type: seq
description: These entities provided funding support for the development of this resource
diff --git a/bin/schema-topic.yaml b/bin/schema-topic.yaml
index f3d36b8a71556..2f96b1a51290e 100644
--- a/bin/schema-topic.yaml
+++ b/bin/schema-topic.yaml
@@ -66,6 +66,11 @@ mapping:
The image ID for an image which contains all of the tools and data for this topic.
_examples:
- quay.io/galaxy/sequence-analysis-training
+ toc:
+ type: bool
+ required: false
+ description:
+ For large topics with many subtopics, set this to true to generate a table of contents above the tutorial table to support quickly jumping to a subtopic.
subtopics:
type: seq
required: false
@@ -219,7 +224,7 @@ mapping:
type: str
description: |
The alt text for the logo (MANDATORY).
- learning_path_cta:
+ learning_path_cta:
type: str
description: |
The specific learning path you wish to reference as a call-to-action for views who aren't sure where to get started.
diff --git a/bin/schema-tutorial.yaml b/bin/schema-tutorial.yaml
index 2c5d8547b85e6..8c618393fa12b 100644
--- a/bin/schema-tutorial.yaml
+++ b/bin/schema-tutorial.yaml
@@ -154,6 +154,13 @@ mapping:
- CONTRIBUTORS
- ORGANISATIONS
- GRANTS
+ reviewing:
+ type: seq
+ description: This person reviewed this material for accuracy and correctness
+ sequence:
+ - type: str
+ enum:
+ - CONTRIBUTORS
funding:
type: seq
description: These entities provided funding support for the development of this resource
diff --git a/events/2022-07-08-gat.md b/events/2022-07-08-gat.md
index 6f4dd17d32a19..b986234f4c5cc 100644
--- a/events/2022-07-08-gat.md
+++ b/events/2022-07-08-gat.md
@@ -119,7 +119,7 @@ program:
topic: admin
- name: tool-integration
topic: dev
- - name: processing-many-samples-at-once
+ - name: collections
topic: galaxy-interface
- name: upload-rules
topic: galaxy-interface
diff --git a/events/2023-04-17-gat-gent.md b/events/2023-04-17-gat-gent.md
index 145886c08bb5a..bea47e80fb06e 100644
--- a/events/2023-04-17-gat-gent.md
+++ b/events/2023-04-17-gat-gent.md
@@ -223,7 +223,7 @@ program:
time: "11:30 - 11:45"
- name: tool-integration
topic: dev
- - name: processing-many-samples-at-once
+ - name: collections
topic: galaxy-interface
- name: upload-rules
topic: galaxy-interface
diff --git a/faqs/galaxy/request-galaxy-tools-on-a-specific-server.md b/faqs/galaxy/request-galaxy-tools-on-a-specific-server.md
index 913656f76c646..5ec359631afc1 100644
--- a/faqs/galaxy/request-galaxy-tools-on-a-specific-server.md
+++ b/faqs/galaxy/request-galaxy-tools-on-a-specific-server.md
@@ -8,8 +8,8 @@ contributors: [nomadscientist]
---
To request tools that already exist in the [Galaxy toolshed](https://toolshed.g2.bx.psu.edu), but not in your server, please raise an issue at:
-- *Europe - usegalaxy.eu* | https://github.com/usegalaxy-eu/usegalaxy-eu-tools
+- *Europe - usegalaxy.eu* \| https://github.com/usegalaxy-eu/usegalaxy-eu-tools
-- *USA - usegalaxy.org* | https://github.com/galaxyproject/usegalaxy-tools
+- *USA - usegalaxy.org* \| https://github.com/galaxyproject/usegalaxy-tools
-- *Australia - usegaalxy.org.au* | https://github.com/usegalaxy-au/usegalaxy-au-tools/tree/master/usegalaxy.org.au
+- *Australia - usegaalxy.org.au* \| https://github.com/usegalaxy-au/usegalaxy-au-tools/tree/master/usegalaxy.org.au
diff --git a/faqs/galaxy/visualisations_igv.md b/faqs/galaxy/visualisations_igv.md
index 7f7119795b9a8..b0b654ed0f723 100644
--- a/faqs/galaxy/visualisations_igv.md
+++ b/faqs/galaxy/visualisations_igv.md
@@ -4,6 +4,7 @@ area: visualisation
box_type: tip
layout: faq
contributors: [shiltemann]
+redirect_from: [/topics/galaxy-interface/tutorials/processing-many-samples-at-once/faqs/visualisations_igv]
---
You can send data from your Galaxy history to IGV for viewing as follows:
diff --git a/learning-pathways/admin-training.md b/learning-pathways/admin-training.md
index 46d484b612419..436cc5c9110f3 100644
--- a/learning-pathways/admin-training.md
+++ b/learning-pathways/admin-training.md
@@ -88,7 +88,7 @@ pathway:
topic: admin
- name: tool-integration
topic: dev
- - name: processing-many-samples-at-once
+ - name: collections
topic: galaxy-interface
- name: upload-rules
topic: galaxy-interface
diff --git a/learning-pathways/climate-learning.md b/learning-pathways/climate-learning.md
index b38075e27078f..83f910e283c18 100644
--- a/learning-pathways/climate-learning.md
+++ b/learning-pathways/climate-learning.md
@@ -1,9 +1,11 @@
---
layout: learning-pathway
-title: Discovering galaxy through climate analysis
+title: Discovering Galaxy through climate analysis
description: |
How to have a complete overview of how Galaxy works going from the user welcome page to use batch tools and finishing by conducting interactive analysis. These set of 3 Climate tutorials allow you to understand and see plenty of the multiple features of Galaxy and learning about the cool subject of climate analysis.
+cover-image: assets/images/galaxy_climate.png
+cover-image-alt: Image of the earth surrounded by a bar colors representing Earth's increasing temperatures.
tags: [Climate, Overview]
editorial_board:
diff --git a/learning-pathways/dev_tools_training.md b/learning-pathways/dev_tools_training.md
new file mode 100644
index 0000000000000..be2c159d93319
--- /dev/null
+++ b/learning-pathways/dev_tools_training.md
@@ -0,0 +1,50 @@
+---
+layout: learning-pathway
+title: Tool development for a nice & shiny subdomain
+type: admin-dev
+description: |
+ Discover Galaxy's communities and learn how to create your subdomain and enrich it by writing, testing and submiting your tools on Galaxy. This learning pathway
+ will guide you through all the steps required to build a tool for Galaxy with Planemo for batch tools and how write an interactive tool.
+cover-image: assets/images/galaxy_subdomain.png
+cover-image-alt: Image of a researcher or developer on a computer thinking of building a community.
+editorial_board:
+- Marie59
+
+tags: [subdomain, community, tool development, 3-day course]
+
+pathway:
+
+ - section: "Day 1: Set up your subdomain for your community"
+ description: This first part explains how to discover the already existing communities (to avoid replication), how to build your subdomain, and finally how to set up your community
+ tutorials:
+ - name: sig_define
+ topic: community
+ - name: subdomain
+ topic: admin
+ - name: sig_create
+ topic: community
+
+ - section: "Day 2: Build a batch tool"
+ description: This module covers getting your package on Conda, a local Galaxy instance with Planemo, write a Galaxy tool, publish it, and make it visible on a Galaxy server.
+ tutorials:
+ - name: tool-from-scratch
+ topic: dev
+ - name: tools_subdomains
+ topic: community
+ - name: community-tool-table
+ topic: community
+
+ - section: "Day 3: Build an interactive tool"
+ description: |
+ Here we go through how to build a docker image and write the correct wrapper for your interactive tool, and then again make it visible on a Galaxy server.
+ tutorials:
+ - name: interactive-tools
+ topic: dev
+ - name: tools_subdomains
+ topic: community
+
+
+
+---
+
+This learning path covers topics on how to build your subdomain, to enrich it, and create a dynamic community.
diff --git a/metadata/git-mod-3f194fb994dca3cfc1faafee668927ca5175dda1.txt b/metadata/git-mod-d67c1b4a044ea32b3995599aa89f93dc43d18701.txt
similarity index 99%
rename from metadata/git-mod-3f194fb994dca3cfc1faafee668927ca5175dda1.txt
rename to metadata/git-mod-d67c1b4a044ea32b3995599aa89f93dc43d18701.txt
index 2ff1e728264ff..863356e1da425 100644
--- a/metadata/git-mod-3f194fb994dca3cfc1faafee668927ca5175dda1.txt
+++ b/metadata/git-mod-d67c1b4a044ea32b3995599aa89f93dc43d18701.txt
@@ -1,3 +1,56 @@
+GTN_GTN:1732881297
+
+topics/assembly/tutorials/mrsa-nanopore/tutorial.md
+topics/microbiome/tutorials/beer-data-analysis/tutorial.md
+GTN_GTN:1732881138
+
+topics/imaging/tutorials/detection-of-mitoflashes/data-library.yaml
+GTN_GTN:1732866147
+
+_config.yml
+topics/contributing/tutorials/updating_tutorial/tutorial.bib
+topics/contributing/tutorials/updating_tutorial/tutorial.md
+GTN_GTN:1732708676
+
+topics/admin/images/create_subdomain/add_customize_tool.png
+topics/admin/images/create_subdomain/add_gxit.png
+topics/admin/images/create_subdomain/customize_tool.png
+topics/admin/images/create_subdomain/earth_system_subdo_gxit.png
+topics/admin/images/create_subdomain/earth_system_subdo_infra.png
+topics/admin/tutorials/subdomain/faqs/index.md
+topics/admin/tutorials/subdomain/tutorial.md
+GTN_GTN:1732642062
+
+_layouts/tutorial_hands_on.html
+GTN_GTN:1732628295
+
+events/2024-12-10-spoc-write-a-thonv2.md
+GTN_GTN:1732616214
+
+topics/microbiome/tutorials/metagenomics-assembly/tutorial.md
+GTN_GTN:1732616101
+
+metadata/workflowhub.yml
+GTN_GTN:1732549127
+
+_layouts/event.html
+events/2024-12-06-spoc-cofest-2024.md
+GTN_GTN:1732525764
+
+faqs/galaxy/analysis_reporting_issues.md
+topics/admin/tutorials/upgrading/tutorial.md
+GTN_GTN:1732514366
+
+topics/imaging/images/detection-of-mitoflashes/Curve_fitting_results_mitoflash.png
+topics/imaging/images/detection-of-mitoflashes/Workflow_mitoflash.png
+GTN_GTN:1732514351
+
+metadata/shortlinks.yaml
+GTN_GTN:1732514334
+
+metadata/git-mod-3f194fb994dca3cfc1faafee668927ca5175dda1.txt
+metadata/git-pub-3f194fb994dca3cfc1faafee668927ca5175dda1.txt
+metadata/github.yml
GTN_GTN:1732294750
topics/contributing/tutorials/create-new-tutorial-content/faqs/icons_list.md
diff --git a/metadata/git-pub-3f194fb994dca3cfc1faafee668927ca5175dda1.txt b/metadata/git-pub-d67c1b4a044ea32b3995599aa89f93dc43d18701.txt
similarity index 99%
rename from metadata/git-pub-3f194fb994dca3cfc1faafee668927ca5175dda1.txt
rename to metadata/git-pub-d67c1b4a044ea32b3995599aa89f93dc43d18701.txt
index 3da07ed321ccf..032f5bb537dad 100644
--- a/metadata/git-pub-3f194fb994dca3cfc1faafee668927ca5175dda1.txt
+++ b/metadata/git-pub-d67c1b4a044ea32b3995599aa89f93dc43d18701.txt
@@ -1,3 +1,26 @@
+GTN_GTN:1732881138
+
+A topics/imaging/tutorials/detection-of-mitoflashes/data-library.yaml
+GTN_GTN:1732866147
+
+A topics/contributing/tutorials/updating_tutorial/tutorial.bib
+A topics/contributing/tutorials/updating_tutorial/tutorial.md
+GTN_GTN:1732708676
+
+A topics/admin/images/create_subdomain/add_customize_tool.png
+A topics/admin/images/create_subdomain/add_gxit.png
+A topics/admin/images/create_subdomain/customize_tool.png
+A topics/admin/images/create_subdomain/earth_system_subdo_gxit.png
+A topics/admin/images/create_subdomain/earth_system_subdo_infra.png
+A topics/admin/tutorials/subdomain/faqs/index.md
+A topics/admin/tutorials/subdomain/tutorial.md
+GTN_GTN:1732628295
+
+A events/2024-12-10-spoc-write-a-thonv2.md
+GTN_GTN:1732514334
+
+R099 metadata/git-mod-7f6ee502828a7660a00ce43e77e864d7cbada116.txt metadata/git-mod-3f194fb994dca3cfc1faafee668927ca5175dda1.txt
+R099 metadata/git-pub-7f6ee502828a7660a00ce43e77e864d7cbada116.txt metadata/git-pub-3f194fb994dca3cfc1faafee668927ca5175dda1.txt
GTN_GTN:1732121078
A _includes/cta.html
diff --git a/metadata/github.yml b/metadata/github.yml
index 089b5d7685415..81d3ed98c5f23 100644
--- a/metadata/github.yml
+++ b/metadata/github.yml
@@ -19429,11 +19429,6 @@
login: hexylena
reactionGroups: []
reviews:
- - author:
- login: hexylena
- state: PENDING
- submittedAt:
- reactionGroups: []
- author:
login: Dirowa
state: COMMENTED
@@ -40369,11 +40364,6 @@
login: abretaud
reactionGroups: []
reviews:
- - author:
- login: hexylena
- state: PENDING
- submittedAt:
- reactionGroups: []
- author:
login: bgruening
state: COMMENTED
@@ -94321,11 +94311,6 @@
login: abretaud
reactionGroups: []
reviews:
- - author:
- login: hexylena
- state: PENDING
- submittedAt:
- reactionGroups: []
- author:
login: abretaud
state: DISMISSED
@@ -109960,11 +109945,6 @@
login: bgruening
reactionGroups: []
reviews:
- - author:
- login: hexylena
- state: PENDING
- submittedAt:
- reactionGroups: []
- author:
login: nsoranzo
state: APPROVED
@@ -225171,3 +225151,548 @@
title: Update Cached Commit Data
updatedAt: '2024-11-18T06:48:02Z'
url: https://github.com/galaxyproject/training-material/pull/5547
+5541:
+ author:
+ id: MDQ6VXNlcjQ1ODY4Mw==
+ is_bot: false
+ login: hexylena
+ name: Helena
+ closedAt: '2024-11-29T11:54:57Z'
+ createdAt: '2024-11-15T09:30:28Z'
+ files:
+ - path: topics/assembly/tutorials/mrsa-nanopore/tutorial.md
+ additions: 9
+ deletions: 9
+ - path: topics/microbiome/tutorials/beer-data-analysis/tutorial.md
+ additions: 0
+ deletions: 1
+ headRefName: ghoul-uaru
+ headRepository:
+ id: MDEwOlJlcG9zaXRvcnkzODI0MTQ2MQ==
+ name: training-material
+ labels:
+ - assembly
+ - microbiome
+ mergedAt: '2024-11-29T11:54:57Z'
+ mergedBy:
+ id: MDQ6VXNlcjI1NjM4NjU=
+ is_bot: false
+ login: shiltemann
+ name: Saskia Hiltemann
+ reactionGroups: []
+ reviews:
+ - author:
+ login: shiltemann
+ state: APPROVED
+ submittedAt: '2024-11-29T11:54:51Z'
+ reactionGroups: []
+ state: MERGED
+ title: Fix MRSA nanopore tutorial CYOA
+ updatedAt: '2024-11-29T12:05:32Z'
+ url: https://github.com/galaxyproject/training-material/pull/5541
+5566:
+ author:
+ is_bot: true
+ login: app/github-actions
+ closedAt: '2024-11-29T11:52:18Z'
+ createdAt: '2024-11-27T01:10:21Z'
+ files:
+ - path: topics/imaging/tutorials/detection-of-mitoflashes/data-library.yaml
+ additions: 19
+ deletions: 0
+ headRefName: create-pull-request/patch-1732669818
+ headRepository:
+ id: MDEwOlJlcG9zaXRvcnkzODI0MTQ2MQ==
+ name: training-material
+ labels: []
+ mergedAt: '2024-11-29T11:52:18Z'
+ mergedBy:
+ id: MDQ6VXNlcjI1NjM4NjU=
+ is_bot: false
+ login: shiltemann
+ name: Saskia Hiltemann
+ reactionGroups: []
+ reviews:
+ - author:
+ login: shiltemann
+ state: APPROVED
+ submittedAt: '2024-11-29T11:52:10Z'
+ reactionGroups: []
+ state: MERGED
+ title: Add missing data-library.yaml files
+ updatedAt: '2024-11-29T11:52:19Z'
+ url: https://github.com/galaxyproject/training-material/pull/5566
+5559:
+ author:
+ id: MDQ6VXNlcjQ0NjA1NzY5
+ is_bot: false
+ login: nomadscientist
+ name: Wendi Bacon
+ closedAt: '2024-11-29T07:42:27Z'
+ createdAt: '2024-11-22T17:19:37Z'
+ files:
+ - path: _config.yml
+ additions: 1
+ deletions: 0
+ - path: topics/contributing/tutorials/updating_tutorial/tutorial.bib
+ additions: 0
+ deletions: 0
+ - path: topics/contributing/tutorials/updating_tutorial/tutorial.md
+ additions: 178
+ deletions: 0
+ headRefName: update_tutorial_spoc
+ headRepository:
+ id: MDEwOlJlcG9zaXRvcnkyMzQwODM1MDM=
+ name: training-material
+ labels:
+ - template-and-tools
+ - contributing
+ mergedAt: '2024-11-29T07:42:27Z'
+ mergedBy:
+ id: MDQ6VXNlcjQ0NjA1NzY5
+ is_bot: false
+ login: nomadscientist
+ name: Wendi Bacon
+ reactionGroups: []
+ reviews:
+ - author:
+ login: bebatut
+ state: COMMENTED
+ submittedAt: '2024-11-28T08:24:44Z'
+ reactionGroups:
+ - content: HEART
+ users:
+ totalCount: 1
+ - author:
+ login: nomadscientist
+ state: COMMENTED
+ submittedAt: '2024-11-28T16:31:25Z'
+ reactionGroups: []
+ - author:
+ login: nomadscientist
+ state: COMMENTED
+ submittedAt: '2024-11-28T16:37:40Z'
+ reactionGroups: []
+ - author:
+ login: nomadscientist
+ state: COMMENTED
+ submittedAt: '2024-11-28T17:00:27Z'
+ reactionGroups: []
+ - author:
+ login: bebatut
+ state: COMMENTED
+ submittedAt: '2024-11-29T07:42:20Z'
+ reactionGroups: []
+ - author:
+ login: bebatut
+ state: APPROVED
+ submittedAt: '2024-11-29T07:42:26Z'
+ reactionGroups: []
+ state: MERGED
+ title: Tutorial on updating tutorials
+ updatedAt: '2024-11-29T07:42:27Z'
+ url: https://github.com/galaxyproject/training-material/pull/5559
+5552:
+ author:
+ id: MDQ6VXNlcjg0OTE5MjQ4
+ is_bot: false
+ login: Marie59
+ name: Marie Jossé
+ closedAt: '2024-11-27T11:57:56Z'
+ createdAt: '2024-11-21T09:07:07Z'
+ files:
+ - path: topics/admin/images/create_subdomain/add_customize_tool.png
+ additions: 0
+ deletions: 0
+ - path: topics/admin/images/create_subdomain/add_gxit.png
+ additions: 0
+ deletions: 0
+ - path: topics/admin/images/create_subdomain/customize_tool.png
+ additions: 0
+ deletions: 0
+ - path: topics/admin/images/create_subdomain/earth_system_subdo_gxit.png
+ additions: 0
+ deletions: 0
+ - path: topics/admin/images/create_subdomain/earth_system_subdo_infra.png
+ additions: 0
+ deletions: 0
+ - path: topics/admin/tutorials/subdomain/faqs/index.md
+ additions: 3
+ deletions: 0
+ - path: topics/admin/tutorials/subdomain/tutorial.md
+ additions: 152
+ deletions: 0
+ headRefName: subdomain
+ headRepository:
+ id: R_kgDOHyVDiA
+ name: training-material
+ labels:
+ - admin
+ mergedAt: '2024-11-27T11:57:56Z'
+ mergedBy:
+ id: MDQ6VXNlcjg2OTc5OTEy
+ is_bot: false
+ login: mira-miracoli
+ name: Mira
+ reactionGroups:
+ - content: HEART
+ users:
+ totalCount: 2
+ reviews:
+ - author:
+ login: hexylena
+ state: COMMENTED
+ submittedAt: '2024-11-21T10:47:03Z'
+ reactionGroups:
+ - content: HOORAY
+ users:
+ totalCount: 1
+ - author:
+ login: mira-miracoli
+ state: COMMENTED
+ submittedAt: '2024-11-25T09:58:24Z'
+ reactionGroups: []
+ - author:
+ login: Marie59
+ state: COMMENTED
+ submittedAt: '2024-11-25T15:48:56Z'
+ reactionGroups: []
+ - author:
+ login: mira-miracoli
+ state: COMMENTED
+ submittedAt: '2024-11-26T07:45:14Z'
+ reactionGroups: []
+ - author:
+ login: mira-miracoli
+ state: APPROVED
+ submittedAt: '2024-11-26T07:45:39Z'
+ reactionGroups:
+ - content: HOORAY
+ users:
+ totalCount: 1
+ - author:
+ login: bgruening
+ state: APPROVED
+ submittedAt: '2024-11-27T11:01:54Z'
+ reactionGroups: []
+ state: MERGED
+ title: Create a Subdomain
+ updatedAt: '2024-11-27T11:57:56Z'
+ url: https://github.com/galaxyproject/training-material/pull/5552
+5565:
+ author:
+ id: MDQ6VXNlcjQxNDc2Nw==
+ is_bot: false
+ login: SandyRogers
+ name: Sandy Rogers
+ closedAt: '2024-11-26T17:27:42Z'
+ createdAt: '2024-11-26T17:20:14Z'
+ files:
+ - path: _layouts/tutorial_hands_on.html
+ additions: 2
+ deletions: 2
+ headRefName: patch-1
+ headRepository:
+ id: R_kgDOLWbm-A
+ name: training-material
+ labels:
+ - template-and-tools
+ mergedAt: '2024-11-26T17:27:42Z'
+ mergedBy:
+ id: MDQ6VXNlcjQ1ODY4Mw==
+ is_bot: false
+ login: hexylena
+ name: Helena
+ reactionGroups: []
+ reviews:
+ - author:
+ login: hexylena
+ state: APPROVED
+ submittedAt: '2024-11-26T17:27:23Z'
+ reactionGroups: []
+ state: MERGED
+ title: Fixes bibtex formatting issues in training material bibtex export
+ updatedAt: '2024-11-26T17:27:42Z'
+ url: https://github.com/galaxyproject/training-material/pull/5565
+5556:
+ author:
+ id: MDQ6VXNlcjQ0NjA1NzY5
+ is_bot: false
+ login: nomadscientist
+ name: Wendi Bacon
+ closedAt: '2024-11-26T13:38:16Z'
+ createdAt: '2024-11-21T18:13:57Z'
+ files:
+ - path: events/2024-12-10-spoc-write-a-thonv2.md
+ additions: 17
+ deletions: 0
+ headRefName: spoc-writeathon-2
+ headRepository:
+ id: MDEwOlJlcG9zaXRvcnkyMzQwODM1MDM=
+ name: training-material
+ labels:
+ - template-and-tools
+ mergedAt: '2024-11-26T13:38:16Z'
+ mergedBy:
+ id: MDQ6VXNlcjQ0NjA1NzY5
+ is_bot: false
+ login: nomadscientist
+ name: Wendi Bacon
+ reactionGroups: []
+ reviews:
+ - author:
+ login: bgruening
+ state: COMMENTED
+ submittedAt: '2024-11-21T18:25:09Z'
+ reactionGroups: []
+ - author:
+ login: hrhotz
+ state: APPROVED
+ submittedAt: '2024-11-26T13:38:14Z'
+ reactionGroups: []
+ state: MERGED
+ title: Spoc writeathon 2
+ updatedAt: '2024-11-26T13:38:16Z'
+ url: https://github.com/galaxyproject/training-material/pull/5556
+5555:
+ author:
+ id: MDQ6VXNlcjI1Njg5NTI1
+ is_bot: false
+ login: bernt-matthias
+ name: M Bernt
+ closedAt: '2024-11-26T10:16:56Z'
+ createdAt: '2024-11-21T11:36:38Z'
+ files:
+ - path: topics/microbiome/tutorials/metagenomics-assembly/tutorial.md
+ additions: 1
+ deletions: 1
+ headRefName: bernt-matthias-patch-1
+ headRepository:
+ id: MDEwOlJlcG9zaXRvcnkzODI0MTQ2MQ==
+ name: training-material
+ labels:
+ - microbiome
+ mergedAt: '2024-11-26T10:16:56Z'
+ mergedBy:
+ id: MDQ6VXNlcjI1NjM4NjU=
+ is_bot: false
+ login: shiltemann
+ name: Saskia Hiltemann
+ reactionGroups: []
+ reviews:
+ - author:
+ login: shiltemann
+ state: APPROVED
+ submittedAt: '2024-11-26T10:16:49Z'
+ reactionGroups: []
+ state: MERGED
+ title: Fix percentage
+ updatedAt: '2024-11-26T10:17:11Z'
+ url: https://github.com/galaxyproject/training-material/pull/5555
+5564:
+ author:
+ is_bot: true
+ login: app/github-actions
+ closedAt: '2024-11-26T10:15:06Z'
+ createdAt: '2024-11-25T13:27:31Z'
+ files:
+ - path: metadata/workflowhub.yml
+ additions: 1
+ deletions: 0
+ headRefName: create-pull-request/patch-1732541249
+ headRepository:
+ id: MDEwOlJlcG9zaXRvcnkzODI0MTQ2MQ==
+ name: training-material
+ labels: []
+ mergedAt: '2024-11-26T10:15:06Z'
+ mergedBy:
+ id: MDQ6VXNlcjI1NjM4NjU=
+ is_bot: false
+ login: shiltemann
+ name: Saskia Hiltemann
+ reactionGroups: []
+ reviews:
+ - author:
+ login: shiltemann
+ state: APPROVED
+ submittedAt: '2024-11-26T10:14:21Z'
+ reactionGroups: []
+ state: MERGED
+ title: Update WorkflowHub IDs
+ updatedAt: '2024-11-26T10:15:06Z'
+ url: https://github.com/galaxyproject/training-material/pull/5564
+5558:
+ author:
+ id: MDQ6VXNlcjQ0NjA1NzY5
+ is_bot: false
+ login: nomadscientist
+ name: Wendi Bacon
+ closedAt: '2024-11-25T15:38:48Z'
+ createdAt: '2024-11-22T13:39:33Z'
+ files:
+ - path: _layouts/event.html
+ additions: 2
+ deletions: 0
+ - path: events/2024-12-06-spoc-cofest-2024.md
+ additions: 11
+ deletions: 3
+ headRefName: nomadscientist-patch-2
+ headRepository:
+ id: MDEwOlJlcG9zaXRvcnkzODI0MTQ2MQ==
+ name: training-material
+ labels:
+ - template-and-tools
+ mergedAt: '2024-11-25T15:38:47Z'
+ mergedBy:
+ id: MDQ6VXNlcjQ0NjA1NzY5
+ is_bot: false
+ login: nomadscientist
+ name: Wendi Bacon
+ reactionGroups: []
+ reviews:
+ - author:
+ login: pavanvidem
+ state: COMMENTED
+ submittedAt: '2024-11-25T13:59:02Z'
+ reactionGroups: []
+ - author:
+ login: nomadscientist
+ state: COMMENTED
+ submittedAt: '2024-11-25T14:09:39Z'
+ reactionGroups: []
+ - author:
+ login: shiltemann
+ state: APPROVED
+ submittedAt: '2024-11-25T15:38:46Z'
+ reactionGroups: []
+ state: MERGED
+ title: Update CoFest
+ updatedAt: '2024-11-25T15:38:48Z'
+ url: https://github.com/galaxyproject/training-material/pull/5558
+5563:
+ author:
+ id: MDQ6VXNlcjM1NDcwOTIx
+ is_bot: false
+ login: emmanuel-ferdman
+ name: Emmanuel Ferdman
+ closedAt: '2024-11-25T09:09:24Z'
+ createdAt: '2024-11-25T08:20:55Z'
+ files:
+ - path: faqs/galaxy/analysis_reporting_issues.md
+ additions: 1
+ deletions: 1
+ - path: topics/admin/tutorials/upgrading/tutorial.md
+ additions: 4
+ deletions: 4
+ headRefName: main
+ headRepository:
+ labels:
+ - admin
+ - faqs
+ mergedAt: '2024-11-25T09:09:24Z'
+ mergedBy:
+ id: MDQ6VXNlcjQ1ODY4Mw==
+ is_bot: false
+ login: hexylena
+ name: Helena
+ reactionGroups: []
+ reviews:
+ - author:
+ login: hexylena
+ state: APPROVED
+ submittedAt: '2024-11-25T09:03:41Z'
+ reactionGroups: []
+ state: MERGED
+ title: Update security guide reference
+ updatedAt: '2024-11-25T09:09:24Z'
+ url: https://github.com/galaxyproject/training-material/pull/5563
+5560:
+ author:
+ is_bot: true
+ login: app/github-actions
+ closedAt: '2024-11-25T05:59:27Z'
+ createdAt: '2024-11-24T23:30:08Z'
+ files:
+ - path: topics/imaging/images/detection-of-mitoflashes/Curve_fitting_results_mitoflash.png
+ additions: 0
+ deletions: 0
+ - path: topics/imaging/images/detection-of-mitoflashes/Workflow_mitoflash.png
+ additions: 0
+ deletions: 0
+ headRefName: create-pull-request/patch-1732491005
+ headRepository:
+ id: MDEwOlJlcG9zaXRvcnkzODI0MTQ2MQ==
+ name: training-material
+ labels: []
+ mergedAt: '2024-11-25T05:59:27Z'
+ mergedBy:
+ id: MDQ6VXNlcjQ2OTk4Mw==
+ is_bot: false
+ login: bgruening
+ name: Björn Grüning
+ reactionGroups: []
+ reviews: []
+ state: MERGED
+ title: Auto Compress Images
+ updatedAt: '2024-11-25T05:59:30Z'
+ url: https://github.com/galaxyproject/training-material/pull/5560
+5561:
+ author:
+ is_bot: true
+ login: app/github-actions
+ closedAt: '2024-11-25T05:59:11Z'
+ createdAt: '2024-11-25T01:10:18Z'
+ files:
+ - path: metadata/shortlinks.yaml
+ additions: 3
+ deletions: 0
+ headRefName: create-pull-request/patch-1732497009
+ headRepository:
+ id: MDEwOlJlcG9zaXRvcnkzODI0MTQ2MQ==
+ name: training-material
+ labels: []
+ mergedAt: '2024-11-25T05:59:11Z'
+ mergedBy:
+ id: MDQ6VXNlcjQ2OTk4Mw==
+ is_bot: false
+ login: bgruening
+ name: Björn Grüning
+ reactionGroups: []
+ reviews: []
+ state: MERGED
+ title: Update Persistent uniform resource locators
+ updatedAt: '2024-11-25T05:59:11Z'
+ url: https://github.com/galaxyproject/training-material/pull/5561
+5562:
+ author:
+ is_bot: true
+ login: app/github-actions
+ closedAt: '2024-11-25T05:58:54Z'
+ createdAt: '2024-11-25T01:11:18Z'
+ files:
+ - path: metadata/git-mod-3f194fb994dca3cfc1faafee668927ca5175dda1.txt
+ additions: 169
+ deletions: 0
+ - path: metadata/git-pub-3f194fb994dca3cfc1faafee668927ca5175dda1.txt
+ additions: 63
+ deletions: 0
+ - path: metadata/github.yml
+ additions: 457
+ deletions: 0
+ headRefName: create-pull-request/patch-1732497076
+ headRepository:
+ id: MDEwOlJlcG9zaXRvcnkzODI0MTQ2MQ==
+ name: training-material
+ labels: []
+ mergedAt: '2024-11-25T05:58:54Z'
+ mergedBy:
+ id: MDQ6VXNlcjQ2OTk4Mw==
+ is_bot: false
+ login: bgruening
+ name: Björn Grüning
+ reactionGroups: []
+ reviews: []
+ state: MERGED
+ title: Update Cached Commit Data
+ updatedAt: '2024-11-25T05:58:58Z'
+ url: https://github.com/galaxyproject/training-material/pull/5562
diff --git a/metadata/shortlinks.yaml b/metadata/shortlinks.yaml
index de0f81b7fb79b..4579ad56adb4b 100644
--- a/metadata/shortlinks.yaml
+++ b/metadata/shortlinks.yaml
@@ -600,6 +600,8 @@ name:
contributing/meta-analysis-plot: "/topics/contributing/tutorials/meta-analysis-plot/tutorial.html"
contributing/running-codespaces: "/topics/contributing/tutorials/running-codespaces/tutorial.html"
imaging/detection-of-mitoflashes: "/topics/imaging/tutorials/detection-of-mitoflashes/tutorial.html"
+ admin/subdomain: "/topics/admin/tutorials/subdomain/tutorial.html"
+ contributing/updating_tutorial: "/topics/contributing/tutorials/updating_tutorial/tutorial.html"
id:
T00000: "/topics/admin/tutorials/ansible/tutorial.html"
T00001: "/topics/admin/tutorials/ansible-galaxy/tutorial.html"
@@ -2067,6 +2069,8 @@ id:
F00437: "/faqs/galaxy/add-toolshed-category-to-a-tool.html"
T00472: "/topics/imaging/tutorials/detection-of-mitoflashes/tutorial.html"
W00291: "/topics/imaging/tutorials/detection-of-mitoflashes/workflows/Capturing-mitoflashes.html"
+ T00473: "/topics/admin/tutorials/subdomain/tutorial.html"
+ T00474: "/topics/contributing/tutorials/updating_tutorial/tutorial.html"
misc:
ansible-galaxy: "/topics/admin/tutorials/ansible-galaxy/tutorial.html"
feeds: "/feeds/index.html"
diff --git a/news/_posts/2024-11-29-tracking-of-mitochondria-and-capturing-mitoflashes.md b/news/_posts/2024-11-29-tracking-of-mitochondria-and-capturing-mitoflashes.md
new file mode 100644
index 0000000000000..db83948cfc0b2
--- /dev/null
+++ b/news/_posts/2024-11-29-tracking-of-mitochondria-and-capturing-mitoflashes.md
@@ -0,0 +1,21 @@
+---
+title: "New Tutorial: Tracking of mitochondria and capturing mitoflashes"
+layout: news
+tags:
+- bioimaging
+- mitoflash
+- mitochondria
+contributions:
+ authorship:
+ - dianichj
+ - kostrykin
+cover: "topics/imaging/images/detection-of-mitoflashes/Workflow_mitoflash.png"
+coveralt: "Analysis Pipeline for Tracking Mitochonria and Capturing Mitoflashes."
+tutorial: topics/imaging/tutorials/detection-of-mitoflashes/tutorial.html
+---
+
+We are happy to share that a new imaging tutorial for **Tracking Mitochondria and Capturing Mitoflashes** is now available in the Galaxy Training Network!
+
+, Mitochondrion tracking (Step 2), and Curve fitting to measured intensities (Step 3).")
+
+This tutorial, Tracking of mitochondria and capturing mitoflashes, provides a step-by-step guide on analyzing mitoflash events in mitochondria using Galaxy. Mitoflashes are transient mitochondrial events characterized by bursts of reactive oxygen species (ROS), membrane potential depolarization, and matrix alkalinization. They are linked to bioenergetic states, physiological processes (like muscle contraction and neuronal development), and may serve as markers of mitochondrial health and longevity. Our tutorial explains how to preprocess imaging data, identify mitoflash events, and quantify key parameters (amplitude, duration, and frequency) using informatics tools in Galaxy. It is designed for researchers studying mitochondrial behavior in health and disease.
diff --git a/news/_posts/2024-12-02-reviewing.md b/news/_posts/2024-12-02-reviewing.md
new file mode 100644
index 0000000000000..2b7e67b5141a4
--- /dev/null
+++ b/news/_posts/2024-12-02-reviewing.md
@@ -0,0 +1,24 @@
+---
+title: "Credit where it's due: GTN Reviewers in the spotlight"
+layout: news
+tags:
+- gtn infrastructure
+- new feature
+- automation
+contributions:
+ authorship:
+ - hexylena
+ - nomadscientist
+ infrastructure:
+ - hexylena
+cover: news/images/reviewing.png
+coveralt: A screenshot of the GTN's short introduction to Galaxy tutorial. There are two authors and two editors, but now shown is a new reviewers with 13 individuals, some overlapping with editors and authors.
+---
+
+We would like to recognise and thank all of the reviewers who have contributed to the GTN tutorials; your efforts are greatly appreciated, and we are grateful for your contributions to the GTN community. Today, we are highlighting your efforts on every single learning material across the GTN.
+
+@gtn:nomadscientist requested the ability to annotate reviewers of a tutorial. What a great idea! We needed a way to give credit to the reviewers who have contributed to the GTN tutorials, as it is usually a somewhat thankless job, there is not a lot of visibility in reviewing code even though it is an incredibly valuable step in the process of developing (e-)learning materials. We quickly [implemented support](https://github.com/galaxyproject/training-material/commit/bce05249d60f571e72c4508da63433b68d243b59) for that contribution role in the GTN infrastructure. Everyone can now manually annotate when a colleague or coworker reviews a tutorial outside of GitHub (as not everyone is familiar or comfortable reviewing learning materials there!)
+
+However given our extensive automation, the we took that one step further! The GTN has recently implemented a new automation that collects metadata about every pull request that is merged into the GTN. This metadata includes the reviewers of learning materials, so of course we can automatically annotate this on every single material within our codebase, leading to our updated headers including up to dozens of previously uncredited reviewers per tutorial.
+
+Thank you all for your hard work and dedication to the GTN community!
diff --git a/news/_posts/2024-12-06-spoc_cofest.md b/news/_posts/2024-12-06-spoc_cofest.md
new file mode 100644
index 0000000000000..cd9f3be5a6194
--- /dev/null
+++ b/news/_posts/2024-12-06-spoc_cofest.md
@@ -0,0 +1,19 @@
+---
+title: "SPOC CoFest 2024: How did it go?"
+contributions:
+ authorship: [nomadscientist]
+tags: [gtn, single-cell]
+layout: news
+cover: "news/images/2023_dec_sc.png"
+coveralt: "swirled cluster dots surround a circle of people all holding hands, looking towards the bright center (future)"
+---
+
+# First SPOC CoFest
+
+We held our first 🖖🏾[SPOC CoFest]({% link events/2024-12-06-spoc-cofest-2024.md %}), in the great tradition of the excellent CoFests organised in the GTN that welcomed @gtn:nomadscientist and many others into the community.
+
+We welcomed a diverse group of participants, from experienced Galaxy trainers to bioinformaticians with little to no Galaxy experience whatsoever. We also were lucky enough to be joined by a 5-strong outpost from Singapore, prompting a Pacific-time-zone kick-off session prior to our day-long event.
+
+🎉A big thank you to all the participants for their hard work!🎉
+
+
diff --git a/news/images/2024-12-06-spoc-cofest_outputs.png b/news/images/2024-12-06-spoc-cofest_outputs.png
new file mode 100644
index 0000000000000..f672f56b00980
Binary files /dev/null and b/news/images/2024-12-06-spoc-cofest_outputs.png differ
diff --git a/news/images/reviewing.png b/news/images/reviewing.png
new file mode 100644
index 0000000000000..d7d2c5bd39b90
Binary files /dev/null and b/news/images/reviewing.png differ
diff --git a/shared/images/nfdi4bioimage.jpeg b/shared/images/nfdi4bioimage.jpeg
new file mode 100644
index 0000000000000..a00128caa8046
Binary files /dev/null and b/shared/images/nfdi4bioimage.jpeg differ
diff --git a/topics/admin/images/create_subdomain/add_customize_tool.png b/topics/admin/images/create_subdomain/add_customize_tool.png
new file mode 100644
index 0000000000000..3ecdacfc3fd60
Binary files /dev/null and b/topics/admin/images/create_subdomain/add_customize_tool.png differ
diff --git a/topics/admin/images/create_subdomain/add_gxit.png b/topics/admin/images/create_subdomain/add_gxit.png
new file mode 100644
index 0000000000000..3a6f5621549c4
Binary files /dev/null and b/topics/admin/images/create_subdomain/add_gxit.png differ
diff --git a/topics/admin/images/create_subdomain/customize_tool.png b/topics/admin/images/create_subdomain/customize_tool.png
new file mode 100644
index 0000000000000..5c745d0682b2d
Binary files /dev/null and b/topics/admin/images/create_subdomain/customize_tool.png differ
diff --git a/topics/admin/images/create_subdomain/earth_system_subdo_gxit.png b/topics/admin/images/create_subdomain/earth_system_subdo_gxit.png
new file mode 100644
index 0000000000000..67ed00bb15569
Binary files /dev/null and b/topics/admin/images/create_subdomain/earth_system_subdo_gxit.png differ
diff --git a/topics/admin/images/create_subdomain/earth_system_subdo_infra.png b/topics/admin/images/create_subdomain/earth_system_subdo_infra.png
new file mode 100644
index 0000000000000..35babf27c4c1e
Binary files /dev/null and b/topics/admin/images/create_subdomain/earth_system_subdo_infra.png differ
diff --git a/topics/admin/tutorials/monitoring/tutorial.md b/topics/admin/tutorials/monitoring/tutorial.md
index 1a8fdc02e49a8..785260c51044b 100644
--- a/topics/admin/tutorials/monitoring/tutorial.md
+++ b/topics/admin/tutorials/monitoring/tutorial.md
@@ -364,7 +364,7 @@ Setting up Telegraf is again very simple. We just add a single role to our playb
> + - plugin: disk
> + - plugin: kernel
> + - plugin: processes
-> + - plugin: io
+> + - plugin: diskio
> + - plugin: mem
> + - plugin: system
> + - plugin: swap
diff --git a/topics/galaxy-interface/tutorials/processing-many-samples-at-once/faqs/index.md b/topics/admin/tutorials/subdomain/faqs/index.md
similarity index 100%
rename from topics/galaxy-interface/tutorials/processing-many-samples-at-once/faqs/index.md
rename to topics/admin/tutorials/subdomain/faqs/index.md
diff --git a/topics/admin/tutorials/subdomain/tutorial.md b/topics/admin/tutorials/subdomain/tutorial.md
new file mode 100644
index 0000000000000..5f99dc42c31f2
--- /dev/null
+++ b/topics/admin/tutorials/subdomain/tutorial.md
@@ -0,0 +1,152 @@
+---
+layout: tutorial_hands_on
+
+title: Create a subdomain for your community on UseGalaxy.eu
+questions:
+- What are the Galaxy communities ?
+- What's a subdomain ?
+- How do I create one ?
+- How can I customize a subdomain ?
+objectives:
+- Discover Galaxy's communities
+- Learn how to set up a subdomain for you community
+- Learn how to customize you tool panel
+- Improve the looks and feel of your subdomain main page
+time_estimation: 1H
+key_points:
+- Subdomain
+- Community
+- Earth System
+tags:
+ - subdomain
+ - community
+ - earth-system
+ - interactive-tools
+contributions:
+ authorship:
+ - Marie59
+ funding:
+ - gallantries
+ - fairease
+ - eurosciencegateway
+
+---
+
+This tutorial covers how to set up a subdomain on usegalaxy.eu. We will take here the example of the [earth system subdomain](https://earth-system.usegalaxy.eu/) and follow the step one by one.
+
+>
+>
+> In this tutorial, we will cover:
+>
+> 1. TOC
+> {:toc}
+>
+{: .agenda}
+
+# Add your subdomain on a Galaxy server
+
+> Subdomain name
+> - Clone the Github repository of the Galaxy server (of where you want your subdomain to be attached)
+> - For Galaxy europe clone the [Infrastructure repo](https://github.com/usegalaxy-eu/infrastructure)
+> - Create a branch on your fork
+> - Open the file **dns.tf** and edit it
+> - You should add the name of your subdomain to the list my_new_and_shiny_subdomain.usegalaxy.eu or for the example here earth-system.usegalaxy.eu AND update **count** (number of subdomains) as shown below.
+>
+> 
+>
+> - Then commit your changes and write a nice message for the admin when you open your Pull Request.
+{: .hands_on}
+
+
+# Enable the interactive tools
+
+> Galaxy Interactive Tool
+> - Clone the Github repository of the Galaxy server (of where you want your subdomain to be attached)
+> - For Galaxy europe clone the [Infrastructure-playbook repo](https://github.com/usegalaxy-eu/infrastructure-playbook/tree/master)
+> - Create a branch on your fork
+> - Open the file **sn06.yml** and edit it
+> - You should add the name of your subdomain to the list "*.my_new_and_shiny_subdomain.usegalaxy.eu" or for the example here "*.earth-system.usegalaxy.eu"
+> - AND add "*.interactivetoolentrypoint.interactivetool.earth-system.usegalaxy.eu" as shown below.
+>
+> 
+>
+> - Then commit your changes and write a nice message for the admin when you open your Pull Request.
+{: .hands_on}
+
+> Add the interactive tool section to the tool panel
+> - First, on your **Infrastructure-playbook** fork, create a new branch
+> - Go to **templates/galaxy/config/**
+> - Open the file **global_host_filters.py.j2** and edit it
+> - In the function per_host_tool_labels add your subdomain as followed
+> ```
+> if "earth-system.usegalaxy.eu" in host:
+> return label.id in valid_labels"
+> ```
+> - Then in the **DOMAIN_SECTIONS** add the the interactive tool section (this will allow to see on the subdomain interface the tool section "Interactive tools") as folowed:
+> `'earth-system': ["interactivetools"],`
+>
+> 
+>
+> - Second, on the same fork go to **files/traefik/rules/**
+> - Open and edit **template-subdomains.yml**, there you need to add the folowing line for your subdomain `{{template "subdomain" "my_new_and_shiny_subdomain"}}`
+>
+> - Finally, commit all your changes and write a nice message for the admin when you open your Pull Request.
+{: .hands_on}
+
+
+# Make a nice tool panel (only for interactive tools)
+
+> Customize the tool panel (only for the interactive tools)
+> - On your **Infrastructure-playbook** fork (don't forget to synchronize it)
+> - First, go to **templates/galaxy/config/**
+> - Open the file **tool_conf.xml.j2** and edit it
+> - From there you can create your own tool sections where your different interactive tools can be organized. See the example of earth-system below.
+> 
+>
+> - Then, commit your changes
+>
+> - Secondly, still in **templates/galaxy/config/**
+> - Open the file **global_host_filters.py.j2** and edit it
+> - Add the different new sections of tools created to your subdomain and prevent those new sections to appear in any other subdomain as shown below.
+> 
+>
+> - Then commit your changes and write a nice message for the admin when you open your Pull Request.
+{: .hands_on}
+
+# Customize the front page
+
+> Update the homepage
+> - For Galaxy europe clone the [website repo](https://github.com/usegalaxy-eu/website/tree/master)
+> - Create a branch on your fork
+> - Create an index file in markdown for instance "index-earth-system.md"
+> - Once created let your imagnination flows to make the looks and feel of your homepage (if needed here is a [PR example](https://github.com/usegalaxy-eu/website/pull/1149))
+> - If you want to add images to your front page add them as folowed **assets/media/my_incredidle_image.png**
+>
+> - Then commit your changes, write a nice message for the admin when you open your Pull Request.
+> If in the future you want to change the front page just update the index file.
+{: .hands_on}
+
+> Add a custom welcome.html
+> There is also the possibility to add a custom welcome.html, as well as other custom static files like the [singlecell subdomain did](https://github.com/usegalaxy-eu/infrastructure-playbook/tree/master/files/galaxy/subdomains/singlecell/static)
+{: .tip}
+
+> Add a theme
+> - On your **Infrastructure-playbook** fork (don't forget to synchronize it)
+> - Go to **group_vars/sn06/**
+> - Open the file **subdomains.yml** and edit it. There you have to add the name of your subdomain.
+> - In this same file you can also customize a theme for your subdomain.
+{: .hands_on}
+
+# Let people know
+
+> Make some communication on your new subdomain
+> - Clone the [galaxy-hub repo](https://github.com/galaxyproject/galaxy-hub/tree/master)
+> - Create a branch on your fork
+> - In **content/news** create a folder for your news example "2023-10-17-earth-system" and in it create an **index.md** file
+> - In this index file write your blog post don't hesitate to add some nice photos
+>
+> - Then commit your changes and open your Pull Request.
+{: .hands_on}
+
+# Conclusion
+Your Subdomain is ready to be used !
diff --git a/topics/assembly/images/image10.png b/topics/assembly/images/image10.png
index 73edc6458dd1a..4e1146ea96c1b 100644
Binary files a/topics/assembly/images/image10.png and b/topics/assembly/images/image10.png differ
diff --git a/topics/assembly/tutorials/mrsa-nanopore/tutorial.md b/topics/assembly/tutorials/mrsa-nanopore/tutorial.md
index eb9f2e0b43cc5..f87381237916d 100644
--- a/topics/assembly/tutorials/mrsa-nanopore/tutorial.md
+++ b/topics/assembly/tutorials/mrsa-nanopore/tutorial.md
@@ -22,8 +22,6 @@ tags:
- nanopore
- assembly
- amr
-- gmod
-- jbrowse1
- microgalaxy
edam_ontology:
- topic_0196 # Sequence Assembly
@@ -153,7 +151,9 @@ The dataset is a FASTQ file.
{% include _includes/cyoa-choices.html option1="Without Illumina MiSeq data" option2="With Illumina MiSeq data" default="Without Illumina MiSeq data" text="Do you have associated Illumina MiSeq data?" disambiguation="miseq"%}
-
+
+
+
> Illumina Data upload
> 1. {% tool [Import](upload1) %} the files from [Zenodo]({{ page.zenodo_link }}) or from the shared data library
@@ -197,7 +197,7 @@ FastQC combines quality statistics from all separate reads and combines them in

-
+
Here, we are going to trim the Illumina data using **fastp** ({% cite Chen2018 %}):
@@ -216,7 +216,7 @@ Here, we are going to trim the Illumina data using **fastp** ({% cite Chen2018 %
> - In *"Read Modification Options"*:
> - In *"Per read cuitting by quality options"*:
> - *Cut by quality in front (5')*: `Yes`
-> - *Cut by quality in front (3')*: `Yes`
+> - *Cut by quality in tail (3')*: `Yes`
> - *Cutting window size*: `4`
> - *Cutting mean quality*: `20`
> - In *"Output Options"*:
@@ -228,7 +228,7 @@ Here, we are going to trim the Illumina data using **fastp** ({% cite Chen2018 %
Depending on the analysis it could be possible that a certain quality or length is needed. The reads can be filtered using the [Filtlong](https://github.com/rrwick/Filtlong) tool. In this training all reads below 1000bp will be filtered.
-
+
When Illumina reads are available, we can use them **if they are good Illumina reads (high depth and complete coverage)** as external reference. In this case, Filtlong ignores the Phred quality scores and instead judges read quality using k-mer matches to the reference (a more accurate gauge of quality).
@@ -240,7 +240,7 @@ When Illumina reads are available, we can use them **if they are good Illumina r
> - In *"Output thresholds"*:
> - *"Min. length"*: `1000`
>
->
+>
> - In *"External references"*:
> - {% icon param-file %} *"Reference Illumina read"*: **fastp** `Read 1 output`
> - {% icon param-file %} *"Reference Illumina read"*: **fastp** `Read 2 output`
@@ -431,7 +431,7 @@ QUAST outputs assembly metrics as an HTML file with metrics and graphs.
> {: .solution}
{: .question}
-
+
## Assembly Polishing
@@ -513,4 +513,4 @@ GC (%) | 32.91 | 32.84
# Conclusion
-In this tutorial, we prepared long reads (using short reads if we had some) assembled them, inspect the produced assembly for its quality, and polished it (if short reads where provided). The assembly, even if uncomplete, is reasonable good to be used in downstream analysis, like [AMR gene detection]({% link topics/genome-annotation/tutorials/amr-gene-detection/tutorial.md %})
\ No newline at end of file
+In this tutorial, we prepared long reads (using short reads if we had some) assembled them, inspect the produced assembly for its quality, and polished it (if short reads where provided). The assembly, even if uncomplete, is reasonable good to be used in downstream analysis, like [AMR gene detection]({% link topics/genome-annotation/tutorials/amr-gene-detection/tutorial.md %})
diff --git a/topics/community/faqs/codex.md b/topics/community/faqs/codex.md
new file mode 100644
index 0000000000000..c1c0f1fd95d1d
--- /dev/null
+++ b/topics/community/faqs/codex.md
@@ -0,0 +1,20 @@
+---
+title: How do I add my community to the Galaxy CoDex?
+box_type: tip
+layout: faq
+contributors: [bebatut, nomadscientist]
+---
+
+You need to create a new folder in the `data/community` folder within [Galaxy Codex code source](https://github.com/galaxyproject/galaxy_codex).
+
+> Create a folder for your community
+>
+> 1. If not already done, fork the [Galaxy Codex repository](https://github.com/galaxyproject/galaxy_codex)
+> 2. Go to the `communities` folder
+> 3. Click on **Add file** in the drop-down menu at the top
+> 4. Select **Create a new file**
+> 5. Fill in the `Name of your file` field with: name of your community + `metadata/categories`
+>
+> This will create a new folder for your community and add a categories file to this folder.
+>
+{: .hands_on}
diff --git a/topics/community/images/tool_subdomain/add_interactive_tool.png b/topics/community/images/tool_subdomain/add_interactive_tool.png
new file mode 100644
index 0000000000000..90ae54696026b
Binary files /dev/null and b/topics/community/images/tool_subdomain/add_interactive_tool.png differ
diff --git a/topics/community/images/tool_subdomain/add_section.png b/topics/community/images/tool_subdomain/add_section.png
new file mode 100644
index 0000000000000..a3479edd6084b
Binary files /dev/null and b/topics/community/images/tool_subdomain/add_section.png differ
diff --git a/topics/community/images/tool_subdomain/ecology_yml_tool.png b/topics/community/images/tool_subdomain/ecology_yml_tool.png
new file mode 100644
index 0000000000000..5b2f05dcd8713
Binary files /dev/null and b/topics/community/images/tool_subdomain/ecology_yml_tool.png differ
diff --git a/topics/community/metadata.yaml b/topics/community/metadata.yaml
index 1db31b8c8f706..1a7fc9a65718c 100644
--- a/topics/community/metadata.yaml
+++ b/topics/community/metadata.yaml
@@ -7,6 +7,7 @@ summary: |
This Topic contains resources for active Galaxy community members to organise and manage working in Galaxy.
#docker_image: "quay.io/galaxy/community"
+learning_path_cta: dev_tools_training
editorial_board:
- nomadscientist
diff --git a/topics/dev/tutorials/community-tool-table/images/galaxy_tool_metadata_extractor_pipeline.png b/topics/community/tutorials/community-tool-table/images/galaxy_tool_metadata_extractor_pipeline.png
similarity index 100%
rename from topics/dev/tutorials/community-tool-table/images/galaxy_tool_metadata_extractor_pipeline.png
rename to topics/community/tutorials/community-tool-table/images/galaxy_tool_metadata_extractor_pipeline.png
diff --git a/topics/dev/tutorials/community-tool-table/images/microgalaxy_tools.png b/topics/community/tutorials/community-tool-table/images/microgalaxy_tools.png
similarity index 100%
rename from topics/dev/tutorials/community-tool-table/images/microgalaxy_tools.png
rename to topics/community/tutorials/community-tool-table/images/microgalaxy_tools.png
diff --git a/topics/dev/tutorials/community-tool-table/tutorial.md b/topics/community/tutorials/community-tool-table/tutorial.md
similarity index 83%
rename from topics/dev/tutorials/community-tool-table/tutorial.md
rename to topics/community/tutorials/community-tool-table/tutorial.md
index 7e708ec8c935e..d54adbed6c5c9 100644
--- a/topics/dev/tutorials/community-tool-table/tutorial.md
+++ b/topics/community/tutorials/community-tool-table/tutorial.md
@@ -2,7 +2,9 @@
layout: tutorial_hands_on
title: Creation of an interactive Galaxy tools table for your community
level: Introductory
-subtopic: tooldev
+redirect_from:
+- /topics/dev/tutorials/community-tool-table/tutorial
+
questions:
- Is it possible to have an overview of all Galaxy tools for a specific scientific domain?
- How can I create a new overview for a specific Galaxy community or domain?
@@ -20,16 +22,17 @@ tags:
contributions:
authorship:
- bebatut
+ - paulzierep
---
-Galaxy offers thousands of tools. They are developed across various GitHub repositories. Furthermore, Galaxy also embraces granular implementation of software tools as sub-modules. In practice, this means that tool suites are separated into Galaxy tools, also known as wrappers, that capture their component operations. Some key examples of suites include [Mothur](https://bio.tools/mothur) and [OpenMS](https://bio.tools/openms), which translate to tens and even hundreds of Galaxy tools.
+Galaxy offers thousands of tools. They are developed across various GitHub repositories. Furthermore, Galaxy also embraces granular implementation of software tools as sub-modules. In practice, this means that tool suites are separated into Galaxy tools, also known as wrappers, that capture their component operations. Some key examples of suites include [Mothur](https://bio.tools/mothur) and [OpenMS](https://bio.tools/openms), which translate to tens and even hundreds of Galaxy tools.
-While granularity supports the composability of tools into rich domain-specific workflows, this decentralized development and sub-module architecture makes it **difficult for Galaxy users to find and reuse tools**. It may also result in Galaxy tool developers **duplicating efforts** by simultaneously wrapping the same software. This is further complicated by a lack of tool metadata, which prevents filtering for all tools in a specific research community or domain, and makes it all but impossible to employ advanced filtering with ontology terms and operations like [EDAM ontology](https://edamontology.org/page).
+While granularity supports the composability of tools into rich domain-specific workflows, this decentralized development and sub-module architecture makes it **difficult for Galaxy users to find and reuse tools**. It may also result in Galaxy tool developers **duplicating efforts** by simultaneously wrapping the same software. This is further complicated by a lack of tool metadata, which prevents filtering for all tools in a specific research community or domain, and makes it all but impossible to employ advanced filtering with ontology terms and operations like [EDAM ontology](https://edamontology.org/page).
The final challenge is also an opportunity: the global nature of Galaxy means that it is a big community. Solving the visibility of tools across this ecosystem and the potential benefits are far-reaching for global collaboration on tool and workflow development.
-To provide the research community with a comprehensive list of available Galaxy tools, [Galaxy Codex](https://github.com/galaxyproject/galaxy_codex) was developed to collect Galaxy wrappers from a list of Git repositories and automatically extract their metadata (including Conda version, [bio.tools](https://bio.tools/) identifiers, and EDAM annotations). The workflow also queries the availability of the tools and usage statistics from the three main Galaxy servers (usegalaxy.*).
+To provide the research community with a comprehensive list of available Galaxy tools, [Galaxy Codex](https://github.com/galaxyproject/galaxy_codex) was developed to collect Galaxy wrappers from a list of Git repositories and automatically extract their metadata (including Conda version, [bio.tools](https://bio.tools/) identifiers, and EDAM annotations). The workflow also queries the availability of the tools and usage statistics from the three main Galaxy servers (usegalaxy.*).

@@ -37,9 +40,9 @@ The pipeline creates an [interactive table with all tools and their metadata](ht
-The generated community-specific interactive table can be used as it and/or embedded, e.g. into the respective Galaxy Hub page or Galaxy subdomain. This table allows further filtering and searching for fine-grained tool selection.
+The generated community-specific interactive table can be used as it and/or embedded, e.g. into the respective Galaxy Hub page or Galaxy subdomain. This table allows further filtering and searching for fine-grained tool selection.
-The pipeline is **fully automated** and executes on a **weekly** basis. Any research community can apply the pipeline to create a table specific to their community.
+The pipeline is **fully automated** and executes on a **weekly** basis. Any research community can apply the pipeline to create a table specific to their community.
The aim is this tutorial is to create such table for a community.
@@ -52,21 +55,13 @@ The aim is this tutorial is to create such table for a community.
>
{: .agenda}
-# Add your community to the Galaxy Codex pipeline
+# Add your community to the Galaxy CoDex
-To create a table for a community, you first need to create a new folder in the `data/community` folder within [Galaxy Codex code source](https://github.com/galaxyproject/galaxy_codex).
+You first need to make sure that your Community is in the [Galaxy CoDex](https://github.com/galaxyproject/galaxy_codex/tree/main/communities), a central resource for Galaxy communities.
-> Create a folder for your community
->
-> 1. If not already done, fork the [Galaxy Codex repository](https://github.com/galaxyproject/galaxy_codex)
-> 2. Go to the `communities` folder
-> 3. Click on **Add file** in the drop-down menu at the top
-> 4. Select **Create a new file**
-> 5. Fill in the `Name of your file` field with: name of your community + `metadata/categories`
->
-> This will create a new folder for your community and add a categories file to this folder.
->
-{: .hands_on}
+{% snippet topics/community/faqs/codex.md %}
+
+# Add your community to the Galaxy Catalog pipeline
One of the filters for the main community table is based on the tool categories on the [Galaxy ToolShed](https://toolshed.g2.bx.psu.edu/). Only tools in the selected ToolShed categories will be added to the filtered table. As a result, it is recommended to include broad categories.
@@ -84,7 +79,7 @@ One of the filters for the main community table is based on the tool categories
>
> 4. Search on the [Galaxy ToolShed](https://toolshed.g2.bx.psu.edu/) for some of the popular tools in your community
> 5. Open the tool entries on the ToolShed, and note their categories
-> 6. Add any new categories to the `categories` file
+> 6. Add any new categories to the `categories` file
{: .hands_on}
Once you have a list of the ToolShed categories that you wish to keep, you can submit this to Galaxy Codex.
@@ -95,22 +90,22 @@ Once you have a list of the ToolShed categories that you wish to keep, you can s
> 2. Fill in the commit message with something like `Add X community`
> 3. Click on `Create a new branch for this commit and start a pull request`
> 4. Create the pull request by following the instructions
->
+>
{: .hands_on}
-The Pull Request will be reviewed. Make sure to respond to any feedback.
+The Pull Request will be reviewed. Make sure to respond to any feedback.
Once the Pull Request is merged, a table with all tool suites and a short description will be created in `communities//resources/tools_filtered_by_ts_categories.tsv`
# Review the generated table to curate tools
-The generated table will contain all the tools associated with the ToolShed categories that you selected. However, not all of these tools might be interesting for your community.
+The generated table will contain all the tools associated with the ToolShed categories that you selected. However, not all of these tools might be interesting for your community.
Galaxy Codex allows for an additional optional filter for tools, that can be defined by the community curator (maybe that is you!).
The additional filter must be stored in a file called `tools_status.tsv` located in `communities//metadata`. The file must include at least 3 columns (with a header):
1. `Suite ID`
-2. `To keep` indicating whether the tool should be included in the final table (TRUE/FALSE).
+2. `To keep` indicating whether the tool should be included in the final table (TRUE/FALSE).
3. `Deprecated` indicating whether the tool is deprecated (TRUE/FALSE).
Example of the `tools_status.tsv` file:
@@ -125,15 +120,19 @@ To generate this file, we recommend you to use the `tools_filtered_by_ts_categor
> Review tools in your community table
>
-> 1. Download the `tools_filtered_by_ts_categories.tsv` file in `communities//resources/`.
-> 2. Open `tools.tsv` with a Spreadsheet Software
-> 3. Review each line corresponding to a tool
+> 1. Download the `tools.tsv` file in `results/`.
+> 2. Open `tools.tsv` with a Spreadsheet Software.
+> 3. Review each line corresponding to a tool.
>
-> 1. Add `TRUE` to the `To keep` column if the tool should be kept, and `FALSE` if not.
-> 2. Add `TRUE` or `FALSE` also to the `Deprecated` column.
+> You can also just review some tools. Those tools that are not reviewed will have be set to `FALSE` in the `Reviewed` column of the updated table.
+> 1. Change the value in the `Reviewed` column from `FALSE` to `TRUE` (this will be done automatically if an entry of the tool in `tools_status.tsv` exists).
+> 2. Add `TRUE` to the `To keep` column if the tool should be kept, and `FALSE` if not.
+> 3. Add `TRUE` or `FALSE` also to the `Deprecated` column.
+> 4. Copy paste the `Galaxy wrapper id`, `To keep`, `Deprecated` columns in a new table (in that order).
>
-> 5. Export the new table as TSV
-> 6. Submit the TSV as `tools_status.tsv` in your `communities//metadata/` folder.
+> This can also be done using the reference function of your Spreadsheet Software.
+> 5. Export the new table as TSV (without header).
+> 6. Submit the TSV as `tools_status.tsv` in your community folder.
> 7. Wait for the Pull Request to be merged
>
{: .hands_on}
@@ -170,4 +169,3 @@ The interactive table you have created can be embedded in your community page on
# Conclusion
You now have an interactive table with Galaxy tools available for your community, and this table is embedded in a community page.
-
diff --git a/topics/community/tutorials/tools_subdomains/faqs/index.md b/topics/community/tutorials/tools_subdomains/faqs/index.md
new file mode 100644
index 0000000000000..9ce3fe4fce824
--- /dev/null
+++ b/topics/community/tutorials/tools_subdomains/faqs/index.md
@@ -0,0 +1,3 @@
+---
+layout: faq-page
+---
diff --git a/topics/community/tutorials/tools_subdomains/tutorial.md b/topics/community/tutorials/tools_subdomains/tutorial.md
new file mode 100644
index 0000000000000..7a7bf5a3c1267
--- /dev/null
+++ b/topics/community/tutorials/tools_subdomains/tutorial.md
@@ -0,0 +1,92 @@
+---
+layout: tutorial_hands_on
+
+title: Make your tools available on your subdomain
+questions:
+- How can a tool be added in a section ?
+- How can a section be added in a subdomain ?
+objectives:
+- Learn to manage the tool panel of a subdomain for both batch and interactive tools
+time_estimation: "30m"
+key_points:
+- Subdomain
+- Tool panel
+- Interactive tools
+tags:
+ - subdomain
+ - earth-system
+ - interactive-tools
+contributions:
+ authorship:
+ - Marie59
+ funding:
+ - gallantries
+ - fairease
+ - eurosciencegateway
+
+---
+This tutorial explains how to make your brand new tools, once they're published through a Pull Request ([check this tutorial]({%link topics/dev/tutorials/tool-from-scratch/tutorial.md %}) on how to build your tool from scracth), visible on your subdomain. Here we'll follow the example on how to make the tools visible on [Galaxy for earth System](https://earth-system.usegalaxy.eu/). You can also find the explanations on how to add your tools on this [Github page](https://usegalaxy-eu.github.io/operations/subdomains.html).
+
+>
+>
+> In this tutorial, we will cover:
+>
+> 1. TOC
+> {:toc}
+>
+{: .agenda}
+
+# Add your batch tool to the right section
+
+> Tool sections (for batch tools)
+> - Clone the Github repository of the Galaxy server (of where your subdomain to be attached)
+> - For Galaxy Europe, fork the [usegalaxy-eu-tools repo](https://github.com/usegalaxy-eu/usegalaxy-eu-tools)
+> - Create a branch on your fork
+> - Browse through the different yaml files and select the one that correspond to your subdomain for earth system everything is made under the ecology owner. The name of the files corresponds to a toolshed owner or community and a few of those communities with a review system and CI integration gets special trust and auto-updates.
+> - Once in the yaml you can add the section for your tool (you want to know what are the existing tool sections ? Go check the [categories defined here](https://github.com/usegalaxy-eu/infrastructure-playbook/blob/master/templates/galaxy/config/tool_conf.xml.j2))
+>
+> An example on how to fill in the yaml file
+> ```
+> - name: id_of_your_tool
+> owner: choose_the_owner_relative_to_a_github_repo
+> tool_panel_section_label: 'Choose the section where your tool belongs'
+> ```
+>
+> 
+>
+> - Then commit your changes and write a nice message for the admin when you open your Pull Request.
+{: .hands_on}
+
+This part is only to make batch tool visible in your subdomain.
+
+# Add a section of tools to your subdomain
+> Sections for your subdomain
+> - If not already done clone the Galaxy Europe [Infrastructure-playbook repo](https://github.com/usegalaxy-eu/infrastructure-playbook/tree/master)
+> - Create a branch on your fork
+> - Go to the file **global_host_filters.py.j2** in **templates/galaxy/config/**
+> - Open it and edit it the "DOMAIN_SECTIONS" part go to your subdomain line and in the list of section add the one you need for instance `"climate_analysis"`
+>
+> 
+>
+> - Then commit your changes and write a nice message for the admin when you open your Pull Request.
+{: .hands_on}
+
+
+# Add you interactive tool to the right section
+> Interactive tool sections
+> - If not already done clone the Galaxy Europe [Infrastructure-playbook repo](https://github.com/usegalaxy-eu/infrastructure-playbook/tree/master)
+> - Create a branch on your fork
+> - Go to the file **tool_conf.xml.j2** in **templates/galaxy/config/**
+> - Open it and edit it by adding the xml file of the interactive tool to the section interactive tool or any other interactive tool section (to know more on how to create your interactive tool section folow [this tutorial]({%link topics/admin/tutorials/subdomain/tutorial.md %}))
+> - Below an example of how to add the interactive tool panoply to the earth system subdomain.
+>
+> 
+>
+> - Then, commit your changes and write a nice message for the admin when you open your Pull Request.
+{: .hands_on}
+
+If you choose to create a new section for your interactive tool don't forget add this section to your subdomain !
+
+# Conclusion
+
+You can now start to build your workflow on your subdomain !
diff --git a/topics/contributing/faqs/github-fork-gtn.md b/topics/contributing/faqs/github-fork-gtn.md
new file mode 100644
index 0000000000000..903a5e73df0d7
--- /dev/null
+++ b/topics/contributing/faqs/github-fork-gtn.md
@@ -0,0 +1,13 @@
+---
+title: "Forking the GTN repository"
+area: github
+box_type: tip
+layout: faq
+contributors: [hexylena, shiltemann]
+---
+
+- Go on the GitHub repository: [github.com/galaxyproject/training-material](https://github.com/galaxyproject/training-material){: width="50%"}
+- Click on the **Fork** button (top-right corner of the page)
+
+
+
diff --git a/topics/contributing/faqs/github-fork-master-main.md b/topics/contributing/faqs/github-fork-master-main.md
new file mode 100644
index 0000000000000..435b976cd64ea
--- /dev/null
+++ b/topics/contributing/faqs/github-fork-master-main.md
@@ -0,0 +1,29 @@
+---
+title: "Updating the default branch from master to main"
+area: github
+box_type: tip
+layout: faq
+contributors: [hexylena, shiltemann]
+---
+
+If you created your fork a long time ago, the default branch on your fork may still be called **master** instead of **main**
+
+1. Point your browser to your fork of the GTN repository
+ - The url will be `https://github.com//training_material` (replacing with your GitHub username)
+
+2. Check the default branch that is shown (at top left).
+
+ 
+
+
+3. Does it say `main`?
+ - Congrats, nothing to do, **you can skip the rest of these steps**
+
+4. Does it say `master`? Then you need to update it, following the instructions below
+
+5. Go to your fork's settings (Click on the gear icon called "Settings")
+6. Find "Branches" on the left
+7. If it says master you can click on the ⇆ icon to switch branches.
+8. Select `main` (it may not be present).
+9. If it isn't present, use the pencil icon to rename `master` to `main`.
+
diff --git a/topics/contributing/faqs/github-fork-sync.md b/topics/contributing/faqs/github-fork-sync.md
new file mode 100644
index 0000000000000..e5754ed14aa2b
--- /dev/null
+++ b/topics/contributing/faqs/github-fork-sync.md
@@ -0,0 +1,20 @@
+---
+title: "Syncing your Fork of the GTN"
+area: github
+box_type: tip
+layout: faq
+contributors: [hexylena, shiltemann]
+---
+
+Whenever you want to contribute something new to the GTN, it is important to start with an up-to-date branch. To do this, you should always update the main branch of your fork, before creating a so-called *feature branch*, a branch where you make your changes.
+
+1. Point your browser to your fork of the GTN repository
+ - The url will be `https://github.com//training_material` (replacing 'your username' with your GitHub username)
+
+2. You might see a message like "This branch is 367 commits behind galaxyproject/training-material:main." as in the screenshot below.
+
+ 
+
+3. Click the **Sync Fork** button on your fork to update it to the latest version.
+
+4. **TIP:** never work directly on your main branch, since that will make the sync process more difficult. Always create a new branch before committing your changes.
diff --git a/topics/contributing/tutorials/gitpod/tutorial.md b/topics/contributing/tutorials/gitpod/tutorial.md
index a03365a9bbb35..a43cb8e745c7e 100644
--- a/topics/contributing/tutorials/gitpod/tutorial.md
+++ b/topics/contributing/tutorials/gitpod/tutorial.md
@@ -47,41 +47,25 @@ If you are working on your own training materials and want preview them online w
> Setting up GitPod
>
-> 1. **Create a fork** of the GTN GitHub repository
-> - Go on the GitHub repository: [github.com/galaxyproject/training-material](https://github.com/galaxyproject/training-material){: width="50%"}
-> - Click on th Fork button (top-right corner of the page)
-> 
+> 1. **Create a fork** of the [GTN GitHub repository](https://github.com/galaxyproject/training-material)
>
-> > Already have a fork of the GTN?
-> > If you already have a fork, fantastic! But a common issue is that the `main` branch gets outdated, or your fork was from before we renamed the `master` branch to `main`.
-> >
-> > - Start by browsing to your fork in GitHub
-> > - Check the default branch that is shown.
-> > - Does it say `master`? Then you need to update it, following the instructions below
-> >
-> > > changing your default branch from master to main
-> > > 1. Go to your fork's settings (Click on the gear icon called "Settings")
-> > > 2. Find "Branches" on the left
-> > > 3. If it says master you can click on the ⇆ icon to switch branches.
-> > > 4. Select `main` (it may not be present).
-> > > 5. If it isn't present, use the pencil icon to rename `master` to `main`.
-> > > 6. Now you can update it in the next step
-> > {: .tip}
-> >
-> > - Click the **Sync Fork** button on your fork to update it to the latest version
-> >
-> > 
-> {: .tip}
+> {% snippet topics/contributing/faqs/github-fork-gtn.md %}
+>
+> 2. Already have a fork of the GTN? Make sure it is up to date.
+>
+> {% snippet topics/contributing/faqs/github-fork-master-main.md %}
+>
+> {% snippet topics/contributing/faqs/github-fork-sync.md %}
>
-> 2. **Open** your browser and navigate to [gitpod.io/login](https://gitpod.io/login)
+> 3. **Open** your browser and navigate to [gitpod.io/login](https://gitpod.io/login)
> - Note: don't leave the `/login` part of the URL off, it will lead you to a different flavour of GitPod. We are using Gitpod classic
-> 3. **Log in** with GitHub
+> 4. **Log in** with GitHub
> {: width="25%"}
-> 4. Click on **Configure your own repository** under the Workspaces menu
+> 5. Click on **Configure your own repository** under the Workspaces menu
> 
-> 5. Under **Select a repository** choose your fork, e.g. `https://github.com/shiltemann/training-material`
+> 6. Under **Select a repository** choose your fork, e.g. `https://github.com/shiltemann/training-material`
> 
-> 6. Click **continue**
+> 7. Click **continue**
> - This will create an enviroment where you can make changes to the GTN and preview them
> - **Note:** It can take quite some time to start up the first time (15-30 minutes)
> - We can make future starts a lot faster using **prebuilds** (see tip box below), you can configure this now while you wait
diff --git a/topics/contributing/tutorials/running-codespaces/images/codespace-publlish.png b/topics/contributing/tutorials/running-codespaces/images/codespace-publlish.png
new file mode 100644
index 0000000000000..019b792afaeac
Binary files /dev/null and b/topics/contributing/tutorials/running-codespaces/images/codespace-publlish.png differ
diff --git a/topics/contributing/tutorials/running-codespaces/images/codespaces-branch-change1.png b/topics/contributing/tutorials/running-codespaces/images/codespaces-branch-change1.png
new file mode 100644
index 0000000000000..1cd8e4e4bbfd8
Binary files /dev/null and b/topics/contributing/tutorials/running-codespaces/images/codespaces-branch-change1.png differ
diff --git a/topics/contributing/tutorials/running-codespaces/images/codespaces-branch-change2.png b/topics/contributing/tutorials/running-codespaces/images/codespaces-branch-change2.png
new file mode 100644
index 0000000000000..582dd0b695b32
Binary files /dev/null and b/topics/contributing/tutorials/running-codespaces/images/codespaces-branch-change2.png differ
diff --git a/topics/contributing/tutorials/running-codespaces/images/codespaces-commit-plus.png b/topics/contributing/tutorials/running-codespaces/images/codespaces-commit-plus.png
new file mode 100644
index 0000000000000..6841eba3dca3a
Binary files /dev/null and b/topics/contributing/tutorials/running-codespaces/images/codespaces-commit-plus.png differ
diff --git a/topics/contributing/tutorials/running-codespaces/tutorial.md b/topics/contributing/tutorials/running-codespaces/tutorial.md
index f9eeb4b98620c..9cbfae87838e0 100644
--- a/topics/contributing/tutorials/running-codespaces/tutorial.md
+++ b/topics/contributing/tutorials/running-codespaces/tutorial.md
@@ -18,12 +18,14 @@ key_points:
contributions:
authorship:
- shiltemann
+ editing:
+ - teresa-m
---
-If you are working on your own training materials and want preview them online without installing anything on your computer, you can do this using GitHub CodeSpaces! Everybody gets 60 free hours of CodeSpaces per month
+If you are working on your own training materials and want preview them online without installing anything on your computer, you can do this using GitHub CodeSpaces! Everybody gets 60 free hours of CodeSpaces per month.
>
@@ -41,17 +43,27 @@ If you are working on your own training materials and want preview them online w
> Setting up GitPod
>
-> 1. Navigate to the GTN GitHub repository, [github.com/galaxyproject/training-material](https://github.com/galaxyproject/training-material)
+> 1. **Create a fork** of the [GTN GitHub repository](https://github.com/galaxyproject/training-material)
>
-> 2. Click on the green **Code** button
+> {% snippet topics/contributing/faqs/github-fork-gtn.md %}
>
-> 3. At the top, switch to the **CodeSpaces** tab
+> 2. Already have a fork of the GTN? Make sure it is up to date.
+>
+> {% snippet topics/contributing/faqs/github-fork-master-main.md %}
+>
+> {% snippet topics/contributing/faqs/github-fork-sync.md %}
+>
+> 2. **Navigate to your fork** of the GTN
+>
+> 3. Click on the green **Code** button
+>
+> 4. At the top, switch to the **CodeSpaces** tab
> 
>
-> 4. Click on **Create codespace on main**
+> 5. Click on **Create codespace on main**
> - Note: if you switch to a specific branch in GitHub first, you can create a codespace for that branch
>
-> 5. This will setup a [Visual Studio Code](https://code.visualstudio.com/) environment for you
+> 6. This will setup a [Visual Studio Code](https://code.visualstudio.com/) environment for you
> - It may take a couple minutes to finish setting everything up
> - In this environment you can also build the GTN website to preview your changes
> - When everything is ready, you should see something like this:
@@ -178,31 +190,44 @@ When you have finished your changes, it all looks good in the preview, you want
> Comitting changes
+> Before you can commit your changes you have to create a branch. You have two options to preform this task:
+> 1. **Option 1: via the terminal**
+> - Hit ctrl+c if your preview was still running to stop it
+> - Create a new branch, commit your changes, push changes:
+>
+> ```bash
+> git checkout -b fix-title
+> git commit -m "update tutorial title" topics/introduction/tutorials/galaxy-intro-short/tutorial.md
+> git push origin fix-title
+> ```
>
-> First we commit our changes inside the codespace:
-> 1. Go to the "Source Control" icon on the left menu bar (it should have a blue dot on it)
-> 2. You should see your changed file (`tutorial.md`)
-> 
-> 3. Hover over the file name, and **click on the plus* icon* to *stage* it
-> 4. Enter a commit message (e.g. "updated tutorial title)
-> 
-> 5. Click on the green **Commit** button
+> 2. **Option 2: via the web interface**
+> - Create a new branch:
+> - On the bottom-left, click on the branch logo (probably labelled "main")
+> 
+> - Enter `fix-title` as the name for your new branch (at top of window)
+> 
+> - Choose "+ Create new branch..." from the dropdown
+> - Commit changes:
+> - On the left menu, click on the "changed files" tab
+> 
+> - You should see your changed file (`tutorial.md`)
+> - Click on the "+" icon next to the file we edited to *stage changes*
+> 
+> - Enter a commit message (top of window)
+> - Hit the checkmark icon below the massage to commit the changes
+> - Publish changes
+> - Click the cloud button at bottom left to publish your changes
+> 
>
{: .hands_on}
-Next, we will push these changes to a branch/fork. We will do this from outside of our codespace for convenience.
+Next, we will see these changes to on your branch/fork. We will do this from outside of our codespace.
> Pushing changes to GitHub
>
> 1. In your browser (outside of codespaces), navigate to the [GTN GitHub page](https://github.com/galaxyproject/training-material)
-> 2. Click on the green **Code** button again
-> 3. Click on the 3-dots menu to the right of your (randomly generated) codespace name
-> 
-> 4. Choose **Export changes to a branch**
-> - For you, it could be **Export changes to fork**
-> 
-> 5. Once it is done, click **See branch** button
-> - This will take you to the new branch
+> 2. GitHub will helpfully show you any recent branches you've pushed to your fork
> - Click the **Compare & pull request** button to create a PR for your changes
> 
{: .hands_on}
diff --git a/topics/contributing/tutorials/updating_tutorial/tutorial.bib b/topics/contributing/tutorials/updating_tutorial/tutorial.bib
new file mode 100644
index 0000000000000..e69de29bb2d1d
diff --git a/topics/contributing/tutorials/updating_tutorial/tutorial.md b/topics/contributing/tutorials/updating_tutorial/tutorial.md
new file mode 100644
index 0000000000000..240f69f95d7b3
--- /dev/null
+++ b/topics/contributing/tutorials/updating_tutorial/tutorial.md
@@ -0,0 +1,178 @@
+---
+layout: tutorial_hands_on
+
+title: Updating tool versions in a tutorial
+subtopic: getting-started
+priority: 6
+
+questions:
+- How can I update the tool versions in a tutorial in the GTN?
+- What else do these updates impact, and how do I update that for consistency?
+
+objectives:
+- Implement tutorial tool version update on existing GTN material
+
+time_estimation: 1H
+key_points:
+- The GTN has a number of fantastic features that make it a cutting-edge resource.
+- Jumping into existing materials can be daunting, but by following these steps, you can be a contribute to this vibrant community!
+
+contributions:
+ authorship:
+ - nomadscientist
+ editing:
+ - wee-snufkin
+ - bebatut
+
+requirements:
+-
+ type: "internal"
+ topic_name: contributing
+ tutorials:
+ - github-interface-contribution
+
+---
+
+Here, we provide a clear set of instructions for updating a GTN tutorial that uses the Galaxy interface for performing analysis. The GTN, and Galaxy, have a number of features to make it as reproducible and shareable as possible, so navigating what needs to be done for an overall update may feel daunting - but not anymore!
+
+We encourage you to pick a tutorial to try this on, so you can use this tutorial side-by-side.
+
+# Phase 1: Find a tutorial with an outdated workflow
+
+{% include _includes/cyoa-choices.html option1="No" option2="Yes" default="No"
+ text="Do you already have a tutorial and workflow you want to update?" %}
+
+
+
+> Check for an outdated workflow
+>
+> 1. **Find a {% icon hands_on %} Hands-on tutorial** in your training topic of interest (for example, Single-cell)
+> 2. **Select the {% icon workflow %} workflow** from the header
+> 3. **Import the workflow** to your favourite Galaxy server
+>
+> {% snippet faqs/galaxy/workflows_import.md %}
+>
+> 4. Go to the **Workflow** menu and select {% icon galaxy-wf-edit %} **Edit workflow**
+> 5. **Click through the tools** in the workflow and check the {% icon tool-versions %} to see if the tools are outdated.
+{: .hands_on}
+
+If the workflow has tools that are up to date (or very close!), great! That tutorial does not need updating! Try another one!
+
+
+
+
+
+> Import the workflow
+>
+> 1. **Navigate to your target {% icon hands_on %} Hands-on tutorial**
+> 2. **Select the {% icon workflow %} workflow** from the header
+> 3. **Import the workflow** to your favourite Galaxy server
+>
+> {% snippet faqs/galaxy/workflows_import.md %}
+>
+> 4. Go to the **Workflow** menu and select {% icon galaxy-wf-edit %} **Edit workflow**
+{: .hands_on}
+
+
+
+# Phase 2: Check that nobody else is working on this
+
+It's always a good idea to check, just in case!
+
+You can:
+ - Send a message to the [GTN Matrix Channel](https://matrix.to/#/#Galaxy-Training-Network_Lobby:gitter.im) is your quickest way forward.
+ - Search through the [GTN Github Repository](https://github.com/galaxyproject/training-material) for existing draft Pull Requests.
+ - Check with [individual communities](https://galaxyproject.org/community/sig/), who may have their own method of tracking. For example, the [🖖🏾Single-cell & sPatial Omics Community](https://galaxyproject.org/community/sig/singlecell/) have a shared [Click-Up board](https://sharing.clickup.com/9015477668/b/h/5-90152810734-2/557452707486fef) at the time of writing.
+
+# Phase 3: Update the workflow
+
+Now, you will update the workflow to using the latest {% icon tool-versions %} tool versions.
+
+> Update the workflow
+>
+> 1. Select {% icon galaxy-wf-options %} **Workflow Options** from the **Workflow Editor**
+> 2. Select {% icon upgrade_workflow %} **Upgrade workflow** to automatically update the tools.
+> 3. Address any issues that may arise.
+> 4. **Save** the workflow.
+{: .hands_on}
+
+
+# Phase 4: Test & fix the workflow
+
+It's crucial to test the workflow, as often times the outputs will be different due to the new tool versions. It can also transpire that the newer tool versions lead to errors, either because they don't work or because you need to change parameter settings that were previously unavailable or not required. Testing is key!
+
+> Test the workflow
+>
+> 1. Import the input datasets from the tutorial you are upgrading (follow the instructions on the tutorial itself for this).
+> 2. Run your updated workflow on the input datasets.
+> 3. Address any issues that may arise, and **NOTE DOWN** all changes.
+> 4. Ensure the workflow meets workflow best practices using the {% icon galaxy-wf-best-practices %} **Best Practices** button and add your name as a contributor, if not there already.
+>
+> {% snippet faqs/galaxy/workflows_best_practices.md %}
+>
+> 5. Add yourself as an author of the workflow.
+> 6. **Save** the `updated workflow`.
+> 7. Run your `updated workflow` on the input datasets in a fresh history.
+> 7. **Save** this history as an `answer key history` & make your history publicly available in Published Histories.
+>
+> {% snippet faqs/galaxy/histories_sharing.md %}
+>
+{: .hands_on}
+
+# Phase 5: Update the tutorial
+
+> If you run out of time
+> You have already completed a large chunk of work (`updated workflow`+ `answer key history`) to get here, and we don't want to lose it!
+> Updating the tutorial to match the `updated workflow` can be a separate contribution. So if you get to this point and run out of time, please:
+> 1. Create an **issue** on the GTN Github Repository and include shareable links to your `updated workflow` & `answer key history`
+> 2. Message on the GTN Matrix channel with a link to your issue and explaining which tutorial you updated.
+> If, however, you are able to finish the task yourself, please read on!
+{: .warning}
+
+Note that you will need to have done either the [Contributing to the GTN Github tutorial using command line]({% link topics/contributing/tutorials/github-command-line-contribution/tutorial.md %}) or the [Contributing to the GTN Github tutorial using Github Desktop]({% link topics/contributing/tutorials/github-interface-contribution/tutorial.md %}). It's helpful as well to understand the folder structure in the GTN, particularly how images go in an image folder either in a tutorial or in the parent folder. Each `topic` has roughly the following structure:
+
+Each topic has the following structure:
+
+```
+├── README.md
+├── metadata.yaml
+├── images
+├── docker
+│ ├── Dockerfile
+├── slides
+│ ├── index.html
+├── tutorials
+│ ├── tutorial1
+│ │ ├── tutorial.md
+│ │ ├── images
+│ │ ├── slides.html
+│ │ ├── data-library.yaml
+│ │ ├── workflows
+│ │ │ ├── workflow.ga
+```
+
+The `tutorial.md` is what you'll be editing, however you will also at the end upload a `workflow.ga` file and likely some image files.
+
+> Update the tools in the tutorial
+>
+> 1. **Update tool versions**: Each {% icon hands_on %} *Hands-on* step in the tutorial will likely have tool (s) with versions in the text. Update these versions / links to be equivalent to your `updated workflow`.
+> 2. **Update tool instructions**: (Simultaneously) update the text to address any differences arising during the update, i.e. new parameters to set or other changes.
+> 3. **Update images**: Wherever images are used to show tool outputs, these will need updating. Use your final `answer key history` for this.
+> 4. **Update text**: Wherever results are referenced in the tutorial text, update these numbers (and possibly interpretation) to reflect the new `answer key history`.
+> 5. **Update header**: In the metadata at the beginning of the tutorial, there are likely `answer key history` links. Update this by adding the link to your `answer key history`.
+> 6. **Update workflow file**: In the tutorial folder in the `training-material` repository, there is a subfolder titled `workflows`. Download your `updated workflow` as a file and deposit that file there.
+> 7. Check if there are any links to workflows/histories in the tutorial text, and if so, update them.
+> 7. Add your name as a *contributor* to the tutorial as an **editor** in the metadata.
+{: .hands_on}
+
+You may find this tutorial to be a helpful reference in the Markdown content of a GTN tutorial: [Creating content in Markdown]({% link topics/contributing/tutorials/create-new-tutorial-content/tutorial.md %})
+
+# Phase 6: Make a pull request and (optional) update workflow testing
+
+At this point, you're welcome to make your Pull Request for the updated tutorial. However, it is likely to fail linting if the workflow testing is not also updated. This can be tricky, so we'd rather you make the Pull Request with all the work you did than let this stop you.
+
+But if you can make the workflow tests, that would be amazing!
+Here's the tutorial: [Creating workflow tests using Planemo]({% link faqs/gtn/gtn_workflow_testing.md %})
+
+
+{% icon congratulations %} Congratulations! You've made it to the end! Thank you so much for contributing to the sustainability of Galaxy tutorials!
diff --git a/topics/data-science/tutorials/online-resources-protein/tutorial.md b/topics/data-science/tutorials/online-resources-protein/tutorial.md
index 289397f2745b6..9fd77c3f37e2e 100644
--- a/topics/data-science/tutorials/online-resources-protein/tutorial.md
+++ b/topics/data-science/tutorials/online-resources-protein/tutorial.md
@@ -2,7 +2,6 @@
layout: tutorial_hands_on
title: One protein along the UniProt page
level: Introductory
-draft: true
zenodo_link: ''
questions:
- How can you search for proteins using text, gene, or protein names?
diff --git a/topics/dev/tutorials/interactive-tools/tutorial.md b/topics/dev/tutorials/interactive-tools/tutorial.md
index 2a1ee90102a73..3b8776ac541a9 100644
--- a/topics/dev/tutorials/interactive-tools/tutorial.md
+++ b/topics/dev/tutorials/interactive-tools/tutorial.md
@@ -38,6 +38,7 @@ contributors:
- abretaud
# editing
- hexylena
+ - Marie59
---
@@ -110,9 +111,7 @@ visualising data, but if it is possible to provide the same
functionality with a regular tool (e.g. by rendering an HTML file as an output),
then an Interactive Tool might not be necessary.
-If you are sure that a static
-output is not sufficient, then it's time to start building your first
-Interactive Tool!
+If you are sure that a static output is not sufficient, then it's time to start building your first Interactive Tool!
> Interactive tool infrastructure
> Interactive tools require some rather complex infrastructure in order to work! However, most of the infrastructure requirements are taken care of by Galaxy core. As such, wrapping a new GxIT requires only three components:
@@ -158,6 +157,10 @@ cycle.
{: .comment}
+{% include _includes/cyoa-choices.html option1="Application" option2="JupyterLab" default="Application" text="Do you want to build a desktop application or a JupyterLab tool ?" %}
+
+
+
# The application
The application that we will wrap in this tutorial is a simple web tool which
@@ -204,6 +207,8 @@ These are specific to your container; these are required for an R-Shiny containe
## The Dockerfile
+If you need some help to start your Dockerfile you can always get some inspiration from the previous interactive tools built in Galaxy. Go check some of the Dockerfiles, for instance the one for the [QGIS](https://github.com/usegalaxy-eu/docker-qgis) application or for [ODV](https://github.com/bgruening/docker-odv/tree/main).
+
> A brief primer on Docker
> Docker allows an entire application context to be containerized. A typical web application consists of an operating system, installed dependancies, web server configuration, database configuration and, of course, the codebase of the software itself. A Docker container can encapsulate all of these components in a single "image", which can be run on any machine with Docker installed.
>
@@ -335,7 +340,7 @@ next to your Dockerfile.
> {: .tip}
{: .hands_on}
-If you are lucky, you might find an available Docker image for the application you are trying to wrap. However, existing Docker images often require some "tweaking" before they will work as a GxIT. Some example configuration changes are:
+If you are lucky, you might find an available Docker image for the application you are trying to wrap. Some configuration changes can be needed such as:
1. Expose the correct port. The application, Docker and tool XML ports must be aligned!
2. Log output to an external file - useful for debugging.
@@ -434,7 +439,7 @@ our new Docker container as a Galaxy tool.
> > Writing the GxIT tool XML
> >
> > * Refer to the [Galaxy tool XML docs](https://docs.galaxyproject.org/en/latest/dev/schema.html).
-> > * You can take inspiration from [Askomics](https://github.com/galaxyproject/galaxy/blob/dev/tools/interactive/interactivetool_askomics.xml), and other [existing Interactive Tools](https://github.com/galaxyproject/galaxy/blob/dev/tools/interactive).
+> > * You can take inspiration from [Askomics](https://github.com/galaxyproject/galaxy/blob/dev/tools/interactive/interactivetool_askomics.xml), [QGIS](https://github.com/usegalaxy-eu/galaxy/blob/release_24.1_europe/tools/interactive/interactivetool_qgis3_34.xml), [ODV](https://github.com/usegalaxy-eu/galaxy/blob/release_24.1_europe/tools/interactive/interactivetool_odv.xml), and other [existing Interactive Tools](https://github.com/galaxyproject/galaxy/blob/dev/tools/interactive).
> > * Check XML syntax with [xmlvalidation.com](https://www.xmlvalidation.com/) or [w3schools XML validator](https://www.w3schools.com/xml/xml_validator.asp), or use a linter in your code editor.
> > * [planemo lint](https://planemo.readthedocs.io/en/latest/commands/lint.html) can also be used for XML linting. But be aware that `planemo test` won't work.
> > * When it comes to testing and debugging your tool XML, it can be easier to update the XML file directly on your Galaxy server between tests.
@@ -525,6 +530,540 @@ our new Docker container as a Galaxy tool.
> Don't forget to change the image path (see the `$REMOTE` variable above) and the citation to fit your project settings.
{: .hands_on}
+# Additional components
+
+The GxIT that we wrapped in this tutorial was a simple example, and you should now understand what is required to create an Interactive Tool for Galaxy. However, there are a few additional components that can enhance the reliability and user experience of the tool. In addition, more complex applications may require some additional components or workarounds the create the desired experience for the user.
+
+## Run script
+
+In the case of our `Tabulator` application, the run script is simply the R script that renders our Shiny App. It is quite straightforward to call this from our Galaxy tool XML.
+However, some web apps might require more elaborate commands to be run. In this situation, there are several solutions demonstrated in the `` section of [existing GxITs](https://github.com/galaxyproject/galaxy/tree/dev/tools/interactive):
+- [Guacamole Desktop](https://github.com/galaxyproject/galaxy/blob/dev/tools/interactive/interactivetool_guacamole_desktop.xml): application startup with `startup.sh`
+- [HiCBrowser](https://github.com/galaxyproject/galaxy/blob/dev/tools/interactive/interactivetool_hicbrowser.xml): application startup with `supervisord`
+- [AskOmics](https://github.com/galaxyproject/galaxy/blob/dev/tools/interactive/interactivetool_askomics.xml): configuration with Python and Bash scripts, followed by `start_all.sh` to run the application.
+
+## Templated config files
+
+Using the `` section in the tool XML, we can enable complex user configuration for the application by templating a run script or configuration file to be read by the application. In this application, for example, we could use a `` section to template user input into the `app.R` script that runs the application within the Docker container. This could enable the user to customize the layout of the app before launch.
+
+## Reserved environment variables
+
+There are a few environment variables
+that are accessible in the command section of the tool XML - these can be handy when writing your tool script.
+[Check the docs](https://docs.galaxyproject.org/en/latest/dev/schema.html#reserved-variables) for a full reference on the tool XML.
+
+```sh
+$__tool_directory__
+$__root_dir__
+$__user_id__
+$__user_email__
+```
+
+It can also be useful to create and inject environment variables into the tool context. This can be achieved using the `` tag in the tool XML. The [RStudio GxIT](https://github.com/galaxyproject/galaxy/blob/b180b7909dc3fe2750fbc8b90214e201eb276794/tools/interactive/interactivetool_rstudio.xml#L12) again provides an example of this:
+
+```xml
+
+ ${__app__.security.encode_id($jupyter_notebook.history_id)}
+ ${__app__.config.galaxy_infrastructure_url}
+ 8080
+ $__galaxy_url__
+ true
+ true
+
+
+```
+
+## Galaxy history interaction
+
+We have demonstrated how to pass an input file to the Docker container. But what if the application needs to interact with the user's Galaxy history? For example, if the user creates a file within the application. That's where the environment variables created in the tool XML become useful.
+
+> Access histories in R
+> From the [R-Studio GxIT](https://github.com/galaxyproject/galaxy/blob/dev/tools/interactive/interactivetool_rstudio.xml) we can see that there is [an R library](https://github.com/hexylena/rGalaxyConnector) that allows us to interact with Galaxy histories.
+>
+> "The convenience functions `gx_put()` and `gx_get()` are available to you to interact with your current Galaxy history. You can save your workspace with `gx_save()`"
+>
+> Under the hood, this library uses [galaxy_ie_helpers](https://github.com/bgruening/galaxy_ie_helpers) - a Python interface to Galaxy histories written with [BioBlend](https://github.com/galaxyproject/bioblend). You could also use BioBlend directly (or even the Galaxy REST API) if your GxIT requires a more flexible interface than these wrappers provide.
+>
+{: .tip}
+
+
+
+
+
+
+# The JupyterLab
+
+The JupyterLab environment that we will wrap in this tutorial is a simple JupyterLab tool which
+allows the user to upload data in a Jupyter environment, manipulate notebooks, and download
+outputs.
+
+Our example JupyterLab can already be found [online](https://github.com/usegalaxy-eu/docker-copernicus-notebooks) .
+But you can also check another implementation of [JupyterLabs](https://github.com/bgruening/docker-jupyter-notebook/blob/master/Dockerfile).
+In the following sections, we will study how it can be built into a GxIT.
+
+
+>
+>
+> First, let's clone the repository to take a quick look at a basic implementation of a JupyterLab.
+>
+> ```console
+> $ git clone https://github.com/bgruening/docker-jupyter-notebook.git
+> $ cd docker-jupyter-notebook
+>
+> $ tree .
+> ├── Dockerfile
+> ├── LICENSE
+> ├── README.md
+> ├── .
+> ├── .
+> ├── .
+> └── startup.sh
+> ```
+> You'll find a Dockerfile, startup.sh, and a README that will describe how the Dockerfile here for a GxIT works. We encourage you to read this README.
+>
+{: .hands_on}
+
+## The Dockerfile
+
+If you need some help to start your Dockerfile you can always get some inspiration from the previous interactive tools built in Galaxy. Go check some of the Dockerfiles, for instance, the one for the [Copernicus data space ecosystem JupyterLab](https://github.com/usegalaxy-eu/docker-copernicus-notebooks/blob/main/Dockerfile) or for a basic [JupyterLab here](https://github.com/bgruening/docker-jupyter-notebook/blob/master/Dockerfile).
+
+> A brief primer on Docker
+> Docker allows an entire application context to be containerized. A typical web application consists of an operating system, installed dependancies, web server configuration, database configuration and, of course, the codebase of the software itself. A Docker container can encapsulate all of these components in a single "image", which can be run on any machine with Docker installed.
+>
+> **Essentials of Docker:**
+>
+> 1. Write an image recipe as a Dockerfile. This single file selects an OS, installs software, pulls code repositories and copies files from the host machine (your computer).
+> 2. Build the image from your recipe:
+>
+> `docker build -t .`
+> 3. View existing images with
+>
+> `docker image list`
+> 4. Run a container with a specified command:
+>
+> `docker run `
+> 5. View running containers:
+>
+> `docker ps`
+> 6. Stop a running container:
+>
+> `docker stop `
+> 7. Remove a stopped container:
+>
+> `docker container rm `
+> 8. Remove an image:
+>
+> `docker image rm `
+{: .tip}
+
+
+Let's check out [the Dockerfile](https://github.com/usegalaxy-eu/docker-copernicus-notebooks/blob/main/Dockerfile) that we'll use to containerize our JupyterLab.
+
+This container recipe can be used to build a Docker image which can be pushed to a
+container registry in the cloud, ready for consumption by our Galaxy instance:
+
+```dockerfile
+# Jupyter container used for Galaxy copernicus notebooks (+other kernels) Integration
+
+# from 5th March 2021
+FROM jupyter/datascience-notebook:python-3.10
+
+MAINTAINER Björn A. Grüning, bjoern.gruening@gmail.com
+
+ENV DEBIAN_FRONTEND noninteractive
+USER root
+
+RUN apt-get -qq update && \
+ apt-get install -y wget unzip net-tools procps && \
+ apt-get autoremove -y && \
+ apt-get clean && \
+ rm -rf /var/lib/apt/lists/* /tmp/* /var/tmp/*
+
+# Set channels to (defaults) > bioconda > conda-forge
+RUN conda config --add channels conda-forge && \
+ conda config --add channels bioconda
+ #conda config --add channels defaults
+RUN pip install --upgrade pip
+RUN pip install --no-cache-dir bioblend galaxy-ie-helpers
+
+ENV JUPYTER /opt/conda/bin/jupyter
+ENV PYTHON /opt/conda/bin/python
+ENV LD_LIBRARY_PATH /opt/conda/lib/
+
+# Python packages
+RUN conda config --add channels conda-forge && \
+ conda config --add channels bioconda && \
+ conda install --yes --quiet \
+ bash_kernel \
+ ansible-kernel \
+ bioblend galaxy-ie-helpers \
+```
+With those packages installed, you have the bases to have a functional environment in your JupyterLab AND be able to link your JupyterLab to the Galaxy history with the package `bioblend galaxy-ie-helpers`.
+
+Then you can add the package specific to the environment you want to have in your JupyterLab for instance
+
+```
+ # specific sentinel, openeo packages
+ sentinelhub \
+ openeo \
+ # other packages for notebooks
+ geopandas \
+ rasterio \
+ ipyleaflet \
+ netcdf4 \
+ h5netcdf \
+ # Jupyter widgets
+ jupytext && \
+ conda clean -yt && \
+ pip install jupyterlab_hdf \
+ fusets
+
+```
+
+Then, you can add some configurations to have some configuration and a welcome notebook by default (this is generic to all JupyterLab GxITs).
+
+```
+ADD ./startup.sh /startup.sh
+ADD ./get_notebook.py /get_notebook.py
+
+# We can get away with just creating this single file and Jupyter will create the rest of the
+# profile for us.
+RUN mkdir -p /home/$NB_USER/.ipython/profile_default/startup/ && \
+ mkdir -p /home/$NB_USER/.jupyter/custom/
+
+COPY ./ipython-profile.py /home/$NB_USER/.ipython/profile_default/startup/00-load.py
+COPY jupyter_notebook_config.py /home/$NB_USER/.jupyter/
+COPY jupyter_lab_config.py /home/$NB_USER/.jupyter/
+
+ADD ./custom.js /home/$NB_USER/.jupyter/custom/custom.js
+ADD ./custom.css /home/$NB_USER/.jupyter/custom/custom.css
+ADD ./default_notebook.ipynb /home/$NB_USER/notebook.ipynb
+
+```
+
+You can also add your own set of notebooks to guide the user like that:
+
+```
+# Download notebooks
+RUN cd /home/$NB_USER/ && \
+ wget -O notebook-samples.zip https://github.com/eu-cdse/notebook-samples/archive/refs/heads/main.zip && \
+ unzip notebook-samples.zip && \
+ rm /home/$NB_USER/notebook-samples.zip && \
+ mv /home/$NB_USER/notebook-samples-main/geo /home/$NB_USER && \
+ mv /home/$NB_USER/notebook-samples-main/sentinelhub /home/$NB_USER && \
+ mv /home/$NB_USER/notebook-samples-main/openeo /home/$NB_USER && \
+ rm -r /home/$NB_USER/notebook-samples-main
+```
+
+Finally, some general variables of environment:
+
+```
+# ENV variables to replace conf file
+ENV DEBUG=false \
+ GALAXY_WEB_PORT=10000 \
+ NOTEBOOK_PASSWORD=none \
+ CORS_ORIGIN=none \
+ DOCKER_PORT=none \
+ API_KEY=none \
+ HISTORY_ID=none \
+ REMOTE_HOST=none \
+ GALAXY_URL=none
+
+# @jupyterlab/google-drive not yet supported
+
+USER root
+WORKDIR /import
+
+# Start Jupyter Notebook
+CMD /startup.sh
+
+```
+
+And with all this your Dockerfile is ready.
+
+You now need to publish this image on a public repository.
+Anyone can use this Dockerfile to rebuild the image if necessary.
+In any case don't forget to have all the files shown when we cloned this [repository](https://github.com/bgruening/docker-jupyter-notebook/tree/master) next to your Dockerfile.
+
+>
+>
+> Let's start working on this Docker container.
+>
+> 1. Install Docker as described on the [docker website](https://docs.docker.com/engine/install/). Click on your distribution name to get specific information.
+>
+> 2. Now let's use the recipe to build our Docker image.
+>
+> ```sh
+> # Build a container image from our Dockerfile
+> IMAGE_TAG="myimage"
+> LOG_PATH=`pwd` # Create log output in current directory
+> PORT=8765
+> docker build -t $IMAGE_TAG --build-arg LOG_PATH=$LOG_PATH --build-arg PORT=$PORT .
+> ```
+>
+> > Automating the build
+> > While developing the Docker container you may find yourself tweaking and rebuilding the container image many times.
+> > In the GitHub repository linked above, you'll notice that the author has used a `Makefile` to accelerate the build and deploy process.
+> > This allows the developer to simply run `make docker` and `make push_hub` to build and push the container, or `make` to rebuild the container after making changes during development. Check out the `Makefile` to see what commands can be run using `make` in this repository.
+> >
+> {: .tip}
+{: .hands_on}
+
+If you are lucky, you might find an available Docker image for the application you are trying to wrap. Some configuration changes can be needed such as:
+
+
+1. Expose the correct port. The application, Docker and tool XML ports must be aligned!
+2. Log output to an external file - useful for debugging.
+3. Make the application callable from tool `` - this sometimes requires a wrapper script to interface the application inside the container (we'll take a look at this later).
+
+
+
+## Test the image
+
+Before we push our container to the cloud, we should give it a local test run to ensure that it's working correctly on our development machine. Have a play and see how our little web app `works!`
+
+>
+> ```sh
+> # Run our application in the container
+> docker run -it -p 127.0.0.1:8765:$PORT $IMAGE_TAG
+>
+> # Or to save time, take advantage of the Makefile
+> make it
+>
+> # Give it a few moments to start up, and the application should be available
+> # in your browser at http://127.0.0.1:8765
+> ```
+{: .hands_on}
+
+## Push the image
+
+If you are happy with the image, we are ready to push it to a container registry
+to make it accessible to our Galaxy server.
+
+During development, we suggest making an account on
+[Docker Hub](https://hub.docker.com/)
+if you don't have one already. This can be used for hosting container images
+during development.
+[Docker Hub](https://hub.docker.com/)
+has great documentation on creating repositories, authenticating with tokens
+and pushing images.
+
+>
+> ```sh
+> # Set remote tag for your container. This should include your username and
+> # repository name for Docker Hub.
+> REMOTE=/my-first-gxit
+>
+> # Tag your image
+> docker tag $IMAGE_TAG:latest $REMOTE:latest
+>
+> # Authenticate your DockerHub account
+> docker login # >>> Enter username and token for your account
+>
+> # Push the image
+> docker push $REMOTE:latest
+> ```
+>
+> > Production container hosting
+> > For production deployment, the
+> > [Galaxy standard](https://docs.galaxyproject.org/en/latest/admin/special_topics/mulled_containers.html)
+> > for container image hosting is
+> > [Biocontainers](https://biocontainers.pro).
+> > This requires you to
+> > [make a pull request](https://biocontainers-edu.readthedocs.io/en/latest/contributing.html)
+> > against the Biocontainers GitHub repository, so this should only be done when an
+> > image is considered production-ready. You can also push your image to a
+> > repository on
+> > [hub.docker.com](https://hub.docker.com) or
+> > [quay.io](https://quay.io)
+> > but please ensure that it links to a public code repository
+> > (e.g. GitHub) to enable maintenance of the image by the Galaxy community!
+> {: .tip}
+{: .hands_on}
+
+You should now have a container in the cloud, ready for action.
+Check out your repo on Docker Hub and you should find the container image there.
+Awesome!
+
+Now we just need to write a tool XML that will enable Galaxy to pull and run
+our new Docker container as a Galaxy tool.
+
+## The tool XML
+
+>
+>
+> Create a Galaxy tool XML file named `interactivetool_copernicus.xml`. The file is similar to a regular tool XML, but calls on our remote Docker image as a dependency. The tags that we are most concerned with are:
+> - A `` (under the `` tag)
+> - A `` which matches our container
+> - An `` file
+> - The `` section
+>
+> > Writing the tool command
+> >
+> > This step can cause a lot of confusion. Here are a few pointer that you will find critical to understanding the process:
+> > - The `` will be templated by Galaxy
+> > - The templated command will run *inside* the Docker container
+> >
+> {: .comment}
+>
+> > Writing the GxIT tool XML
+> >
+> > * Refer to the [Galaxy tool XML docs](https://docs.galaxyproject.org/en/latest/dev/schema.html).
+> > * You can take inspiration from [Copernicus](https://github.com/usegalaxy-eu/galaxy/blob/release_24.1_europe/tools/interactive/interactivetool_copernicus.xml) or for a more advanced [jupyterlab](https://github.com/usegalaxy-eu/galaxy/blob/release_24.1_europe/tools/interactive/interactivetool_jupyter_notebook_1.0.1.xml) [existing Interactive Tools](https://github.com/galaxyproject/galaxy/blob/dev/tools/interactive).
+> > * Check XML syntax with [xmlvalidation.com](https://www.xmlvalidation.com/) or [w3schools XML validator](https://www.w3schools.com/xml/xml_validator.asp), or use a linter in your code editor.
+> > * [planemo lint](https://planemo.readthedocs.io/en/latest/commands/lint.html) can also be used for XML linting. But be aware that `planemo test` won't work.
+> > * When it comes to testing and debugging your tool XML, it can be easier to update the XML file directly on your Galaxy server between tests.
+> {: .tip}
+>
+> >
+> >
+> > {% raw %}
+> >
+> > ```xml
+> >
+> > sample notebooks to access and discover data
+> >
+> > 0.0.1
+> >
+> >
+> > quay.io/galaxy/copernicus-jupyterlab:@VERSION@
+> >
+> >
+> >
+> > 8888
+> > ipython/lab
+> >
+> >
+> >
+> > $__history_id__
+> > $__galaxy_url__
+> > 8080
+> > $__galaxy_url__
+> > < environment_variable name="API_KEY" inject="api_key" />
+> >
+> > > #import re
+> > export GALAXY_WORKING_DIR=`pwd` &&
+> > mkdir -p ./jupyter/outputs/ &&
+> > mkdir -p ./jupyter/data &&
+> > mkdir -p ./jupyter/notebooks &&
+> > mkdir -p ./jupyter/notebooks/geo &&
+> > mkdir -p ./jupyter/notebooks/openeo &&
+> > mkdir -p ./jupyter/notebooks/sentinelhub &&
+> >
+> > #for $count, $file in enumerate($input):
+> > #set $cleaned_name = str($count + 1) + '_' + re.sub('[^\w\-\.\s]', '_', str($file.element_identifier))
+> > ln -sf '$file' './jupyter/data/${cleaned_name}' &&
+> > #end for
+> >
+> > ## change into the directory where the notebooks are located
+> > cd ./jupyter/ &&
+> > export HOME=/home/jovyan/ &&
+> > export PATH=/home/jovyan/.local/bin:\$PATH &&
+> >
+> > #if $mode.mode_select == 'scratch'
+> > ## copy all notebooks, workflows and data
+> > cp '$__tool_directory__/default_notebook.ipynb' ./ipython_galaxy_notebook.ipynb &&
+> > jupyter trust ./ipython_galaxy_notebook.ipynb &&
+> > cp -r /home/\$NB_USER/geo/* ./notebooks/geo/ &&
+> > cp -r /home/\$NB_USER/openeo/* ./notebooks/openeo/ &&
+> > cp -r /home/\$NB_USER/sentinelhub/* ./notebooks/sentinelhub/ &&
+> >
+> >
+> > ## provide all rights to copied files
+> > jupyter lab --allow-root --no-browser &&
+> > cp ./*.ipynb '$jupyter_notebook' &&
+> >
+> > cd outputs/ &&
+> > sleep 2 &&
+> > for file in *; do mv "\$file" "\$file.\${file\#\#*.}"; done
+> > #else
+> > #set $noteboook_name = re.sub('[^\w\-\.\s]', '_', str($mode.ipynb.element_identifier))
+> > cp '$mode.ipynb' './${noteboook_name}.ipynb' &&
+> > jupyter trust './${noteboook_name}.ipynb' &&
+> > #if $mode.run_it
+> > jupyter nbconvert --to notebook --execute --output ./ipython_galaxy_notebook.ipynb --allow-errors ./*.ipynb &&
+> > #set $noteboook_name = 'ipython_galaxy_notebook'
+> > #else
+> > jupyter lab --allow-root --no-browser --NotebookApp.shutdown_button=True &&
+> > #end if
+> > cp './${noteboook_name}.ipynb' '$jupyter_notebook' &&
+> >
+> > cd outputs/ &&
+> > sleep 2 &&
+> > for file in *; do mv "\$file" "\$file.\${file\#\#*.}"; done
+> > #end if
+> > ]]>
+> >
+> >
+> >
+> >
+> >
+> >
+> >
+> >
+> >
+> >
+> >
+> > > help="This option is useful in workflows when you just want to execute a notebook and not dive into the webfrontend."/>
+> >
+> >
+> >
+> >
+> >
+> >
+> >
+> >
+> >
+> >
+> >
+> >
+> >
+> >
+> >
+> >
+> >
+> >
+> >
+> > This tool contains sample Jupyter notebooks for the Copernicus Data Space Ecosystem. Notebooks are grouped per kernel: sentinelhub, openeo and geo. To know more https:/github.com/eu-cdse/notebook-samples
+> >
+> > To have more example notebooks produced by th OpenEO community that you can use in this jupyterlab go there https://github.com/Open-EO/openeo-community-examples/tree/main
+> >
+> >
+> >
+> >
+> > @Manual{,
+> > title = {Copernicus Data Space Ecosystem },
+> > author = {The eu-cdse community},
+> > year = {2023},
+> > note = {https://github.com/eu-cdse}
+> >
+> >
+> >
+> > ```
+> > {% endraw %}
+> {: .solution}
+>
+> In order to get your data from your galaxy history into your jupyterlab don't forget to have this kind of line in the executable
+> ```
+> #for $count, $file in enumerate($input):
+> #set $cleaned_name = str($count + 1) + '_' + re.sub('[^\w\-\.\s]', '_', str($file.element_identifier))
+> ln -sf '$file' './jupyter/data/${cleaned_name}' &&
+> #end for
+> ```
+> AND to get your output in your history data
+>
+> ```
+>
+>
+>
+> ```
+> Don't forget to change the image path and the citation to fit your project settings.
+{: .hands_on}
+
+
+
# Testing locally
You would like to check your GxIT integration in Galaxy but don't have a development server or don't want to disturb your sysadmin at this point?
@@ -581,6 +1120,7 @@ Let's check this integration on your machine. You can use a VM if you prefer not
> ```xml
>
>
+>
>
> ```
> With these lines, Galaxy will create a new section named "Interactive tools" in the tool panel
@@ -617,10 +1157,14 @@ Let's check this integration on your machine. You can use a VM if you prefer not
>
>
> ```
-> Finally, copy your GxIT wrapper to the Interactive Tool directory:
+> Finally, copy your GxIT wrapper to the Interactive Tool directory (depending on if you followed the Application or Jupyterlab part of the tuto):
> ```sh
> cp ~/my_filepath/interactivetool_tabulator.xml ~/GxIT/galaxy/tools/interactive/
> ```
+> OR
+> ```sh
+> cp ~/my_filepath/interactivetool_copernicus.xml ~/GxIT/galaxy/tools/interactive/
+> ```
{: .hands_on}
## Run Galaxy
@@ -685,6 +1229,14 @@ the Galaxy core application directory, and adding the tool to our
> >
> >
> > ```
+> > OR
+> > ```
+> >
+> >
+> >
+> >
+> >
+> > ```
> {: .solution}
>
> 5. Now we just need to restart the Galaxy server to refresh the tool registry
@@ -734,6 +1286,20 @@ Have a look in the web interface of your Galaxy instance. You should find the ne
> ```
> {% endraw %}
>
+> OR
+>
+> {% raw %}
+> ```xml
+>
+>
+>
+>
+>
+>
+> ```
+> {% endraw %}
+>
+>
> 3. Create variables in the following sections of `group_vars/galaxyservers.yml`
>
> {% raw %}
@@ -768,58 +1334,6 @@ The most obvious way to test a tool is simply to run it in the Galaxy UI, straig
{: .tip}
-# Additional components
-
-The GxIT that we wrapped in this tutorial was a simple example, and you should now understand what is required to create an Interactive Tool for Galaxy. However, there are a few additional components that can enhance the reliability and user experience of the tool. In addition, more complex applications may require some additional components or workarounds the create the desired experience for the user.
-
-## Run script
-In the case of our `Tabulator` application, the run script is simply the R script that renders our Shiny App. It is quite straightforward to call this from our Galaxy tool XML. However, some web apps might require more elaborate commands to be run. In this situation, there are a number of solutions demonstrated in the `` section of [existing GxITs](https://github.com/galaxyproject/galaxy/tree/dev/tools/interactive):
-- [Guacamole Desktop](https://github.com/galaxyproject/galaxy/blob/dev/tools/interactive/interactivetool_guacamole_desktop.xml): application startup with `startup.sh`
-- [HiCBrowser](https://github.com/galaxyproject/galaxy/blob/dev/tools/interactive/interactivetool_hicbrowser.xml): application startup with `supervisord`
-- [AskOmics](https://github.com/galaxyproject/galaxy/blob/dev/tools/interactive/interactivetool_askomics.xml): configuration with Python and Bash scripts, followed by `start_all.sh` to run the application.
-
-## Templated config files
-Using the `` section in the tool XML, we can enable complex user configuration for the application by templating a run script or configuration file to be read by the application. In this application, for example, we could use a `` section to template user input into the `app.R` script that runs the application within the Docker container. This could enable the user to customize the layout of the app before launch.
-
-## Reserved environment variables
-
-There are a few environment variables
-that are accessible in the command section of the tool XML - these can be handy when writing your tool script.
-[Check the docs](https://docs.galaxyproject.org/en/latest/dev/schema.html#reserved-variables) for a full reference on the tool XML.
-
-```sh
-$__tool_directory__
-$__root_dir__
-$__user_id__
-$__user_email__
-```
-
-It can also be useful to create and inject environment variables into the tool context. This can be acheived using the `` tag in the tool XML. The [RStudio GxIT](https://github.com/galaxyproject/galaxy/blob/b180b7909dc3fe2750fbc8b90214e201eb276794/tools/interactive/interactivetool_rstudio.xml#L12) again provides an example of this:
-
-```xml
-
- ${__app__.security.encode_id($jupyter_notebook.history_id)}
- ${__app__.config.galaxy_infrastructure_url}
- 8080
- $__galaxy_url__
- true
- true
-
-
-```
-
-## Galaxy history interaction
-We have demonstrated how to pass an input file to the Docker container. But what if the application needs to interact with the user's Galaxy history? For example, if the user creates a file within the application. That's where the environment variables created in the tool XML become useful.
-
-> Access histories in R
-> From the [R-Studio GxIT](https://github.com/galaxyproject/galaxy/blob/dev/tools/interactive/interactivetool_rstudio.xml) we can see that there is [an R library](https://github.com/hexylena/rGalaxyConnector) that allows us to interact with Galaxy histories.
->
-> "The convenience functions `gx_put()` and `gx_get()` are available to you to interact with your current Galaxy history. You can save your workspace with `gx_save()`"
->
-> Under the hood, this library uses [galaxy_ie_helpers](https://github.com/bgruening/galaxy_ie_helpers) - a Python interface to Galaxy histories written with [BioBlend](https://github.com/galaxyproject/bioblend). You could also use BioBlend directly (or even the Galaxy REST API) if your GxIT requires a more flexible interface than these wrappers provide.
->
-{: .tip}
-
## Self-destruct script
Unlike regular tools that exit after the execution of the underlying command is complete, web applications will run indefinitely until terminated. With Galaxy's legacy "Interactive Environments", this used to result in "zombie" containers hanging around and clogging up the Galaxy server. You may notice a `terminate.sh` script in some older GxITs as a workaround to this problem, but the new GxIT architecture handles container termination for you. This script is no longer required nor recommended.
diff --git a/topics/ecology/tutorials/Metashrimps_tutorial/tutorial.md b/topics/ecology/tutorials/Metashrimps_tutorial/tutorial.md
index ae49d6024dc5b..b7bf1e2f47bd5 100644
--- a/topics/ecology/tutorials/Metashrimps_tutorial/tutorial.md
+++ b/topics/ecology/tutorials/Metashrimps_tutorial/tutorial.md
@@ -86,7 +86,7 @@ publishable as a real Data Paper giving recognition to all the people that helpe
>
> 1. Create a new history for this tutorial
> 2. Import this metadata file from [Zenodo]({{ page.zenodo_link }}) to test it
-> -> Training Data for "Creating Quality FAIR assessment reports and draft of Data Papers from EML metadata with MetaShRIMPS"):
+> -> Training Data for "Creating Quality FAIR assessment reports and draft of Data Papers from EML metadata with MetaShRIMPS":
> ```
> https://zenodo.org/record/8130567/files/Kakila_database_marine_mammal.xml
> ```
@@ -109,7 +109,7 @@ you want to use.
{:width="500"}
-After uploading the file, or if you have indicate it as input data if the tool, you just have to click on **Execute** to launch the tool with the file.
+After uploading the file, or if you have indicated it as input data in the tool form, you just have to click on **Execute** to launch MetaShRIMPS jobs with the file.
# Outputs
diff --git a/topics/fair/tutorials/earth_system_rocrate/faqs/index.md b/topics/fair/tutorials/earth_system_rocrate/faqs/index.md
new file mode 100644
index 0000000000000..9ce3fe4fce824
--- /dev/null
+++ b/topics/fair/tutorials/earth_system_rocrate/faqs/index.md
@@ -0,0 +1,3 @@
+---
+layout: faq-page
+---
diff --git a/topics/fair/tutorials/earth_system_rocrate/images/all_tree_structure.png b/topics/fair/tutorials/earth_system_rocrate/images/all_tree_structure.png
new file mode 100644
index 0000000000000..518986742b6e4
Binary files /dev/null and b/topics/fair/tutorials/earth_system_rocrate/images/all_tree_structure.png differ
diff --git a/topics/fair/tutorials/earth_system_rocrate/images/rocrate1.png b/topics/fair/tutorials/earth_system_rocrate/images/rocrate1.png
new file mode 100644
index 0000000000000..3f87224d5dd30
Binary files /dev/null and b/topics/fair/tutorials/earth_system_rocrate/images/rocrate1.png differ
diff --git a/topics/fair/tutorials/earth_system_rocrate/images/rocrate10.png b/topics/fair/tutorials/earth_system_rocrate/images/rocrate10.png
new file mode 100644
index 0000000000000..a70e11ab7565e
Binary files /dev/null and b/topics/fair/tutorials/earth_system_rocrate/images/rocrate10.png differ
diff --git a/topics/fair/tutorials/earth_system_rocrate/images/rocrate11.png b/topics/fair/tutorials/earth_system_rocrate/images/rocrate11.png
new file mode 100644
index 0000000000000..85130fb1bda8b
Binary files /dev/null and b/topics/fair/tutorials/earth_system_rocrate/images/rocrate11.png differ
diff --git a/topics/fair/tutorials/earth_system_rocrate/images/rocrate12.png b/topics/fair/tutorials/earth_system_rocrate/images/rocrate12.png
new file mode 100644
index 0000000000000..c0b7f8108450d
Binary files /dev/null and b/topics/fair/tutorials/earth_system_rocrate/images/rocrate12.png differ
diff --git a/topics/fair/tutorials/earth_system_rocrate/images/rocrate13.png b/topics/fair/tutorials/earth_system_rocrate/images/rocrate13.png
new file mode 100644
index 0000000000000..eff62d84c950e
Binary files /dev/null and b/topics/fair/tutorials/earth_system_rocrate/images/rocrate13.png differ
diff --git a/topics/fair/tutorials/earth_system_rocrate/images/rocrate2.png b/topics/fair/tutorials/earth_system_rocrate/images/rocrate2.png
new file mode 100644
index 0000000000000..3f9928e1eb290
Binary files /dev/null and b/topics/fair/tutorials/earth_system_rocrate/images/rocrate2.png differ
diff --git a/topics/fair/tutorials/earth_system_rocrate/images/rocrate3.png b/topics/fair/tutorials/earth_system_rocrate/images/rocrate3.png
new file mode 100644
index 0000000000000..1499145647a67
Binary files /dev/null and b/topics/fair/tutorials/earth_system_rocrate/images/rocrate3.png differ
diff --git a/topics/fair/tutorials/earth_system_rocrate/images/rocrate4.png b/topics/fair/tutorials/earth_system_rocrate/images/rocrate4.png
new file mode 100644
index 0000000000000..43a1237fc838a
Binary files /dev/null and b/topics/fair/tutorials/earth_system_rocrate/images/rocrate4.png differ
diff --git a/topics/fair/tutorials/earth_system_rocrate/images/rocrate5.png b/topics/fair/tutorials/earth_system_rocrate/images/rocrate5.png
new file mode 100644
index 0000000000000..6b580eb913cb8
Binary files /dev/null and b/topics/fair/tutorials/earth_system_rocrate/images/rocrate5.png differ
diff --git a/topics/fair/tutorials/earth_system_rocrate/images/rocrate6.png b/topics/fair/tutorials/earth_system_rocrate/images/rocrate6.png
new file mode 100644
index 0000000000000..73cbd87a9c70f
Binary files /dev/null and b/topics/fair/tutorials/earth_system_rocrate/images/rocrate6.png differ
diff --git a/topics/fair/tutorials/earth_system_rocrate/images/rocrate7.png b/topics/fair/tutorials/earth_system_rocrate/images/rocrate7.png
new file mode 100644
index 0000000000000..4407a640a48bf
Binary files /dev/null and b/topics/fair/tutorials/earth_system_rocrate/images/rocrate7.png differ
diff --git a/topics/fair/tutorials/earth_system_rocrate/images/rocrate8.png b/topics/fair/tutorials/earth_system_rocrate/images/rocrate8.png
new file mode 100644
index 0000000000000..7faf37965be9d
Binary files /dev/null and b/topics/fair/tutorials/earth_system_rocrate/images/rocrate8.png differ
diff --git a/topics/fair/tutorials/earth_system_rocrate/images/rocrate9.png b/topics/fair/tutorials/earth_system_rocrate/images/rocrate9.png
new file mode 100644
index 0000000000000..ad51204e4abc2
Binary files /dev/null and b/topics/fair/tutorials/earth_system_rocrate/images/rocrate9.png differ
diff --git a/topics/fair/tutorials/earth_system_rocrate/images/workflow_marino.png b/topics/fair/tutorials/earth_system_rocrate/images/workflow_marino.png
new file mode 100644
index 0000000000000..c0700487cf4c8
Binary files /dev/null and b/topics/fair/tutorials/earth_system_rocrate/images/workflow_marino.png differ
diff --git a/topics/fair/tutorials/earth_system_rocrate/images/workflow_rocrate.png b/topics/fair/tutorials/earth_system_rocrate/images/workflow_rocrate.png
new file mode 100644
index 0000000000000..92eea18c23691
Binary files /dev/null and b/topics/fair/tutorials/earth_system_rocrate/images/workflow_rocrate.png differ
diff --git a/topics/fair/tutorials/earth_system_rocrate/slides.html b/topics/fair/tutorials/earth_system_rocrate/slides.html
new file mode 100644
index 0000000000000..bee5329ab9d45
--- /dev/null
+++ b/topics/fair/tutorials/earth_system_rocrate/slides.html
@@ -0,0 +1,138 @@
+---
+layout: tutorial_slides
+logo: "GTN"
+
+title: "An earth-system RO-Crate"
+video: true
+questions:
+ - "How to generate a RO-Crate"
+ - "How to follow a Marine Omics study"
+ - "How to read a RO-Crate ?"
+objectives:
+ - "Understand RO-Crate in an earth-system context"
+time_estimation: 1H
+tags:
+ - earth-system
+ - ro-crate
+ - ocean
+ - marine omics
+key_points:
+ - "RO-Crate in earth-system analysis"
+ - "Marine omics workflow "
+contributions:
+ authorship:
+ - Marie59
+ funding:
+ - fairease
+ - eurosciencegateway
+---
+
+# Workflow context
+
+The workflow used as an example here goes from :
+- First, produce a protein FASTA file from a nucleotide FASTA file using ({% tool [Prodigal](tool_id=toolshed.g2.bx.psu.edu/repos/iuc/prodigal/prodigal/2.6.3+galaxy0) %}) (a tool that predicts protein-coding genes from DNA sequences).
+- Then, use ({% tool [InterProscan](toolshed.g2.bx.psu.edu/repos/bgruening/interproscan/interproscan/5.59-91.0+galaxy3) %}) to create a tabular. Interproscan is a batch tool to query the InterPro database. It helps identify and predict the functions of proteins by comparing them to known databases.
+- Finally, discover ({% tool [SanntiS](toolshed.g2.bx.psu.edu/repos/ecology/sanntis_marine/sanntis_marine/0.9.3.5+galaxy1) %}) both to build genbank and especially to detect and annotate biosynthetic gene clusters (BGCs).
+---
+
+## The workflow itself
+
+
+---
+
+## Run the workflow
+
+Import and run the [Biosynthetic Gene Clusters workflow](https://earth-system.usegalaxy.eu/u/marie.josse/w/marine-omics-identifying-biosynthetic-gene-clusters)
+
+To know more on how to do that follow the [tutorial]({% link topics/ecology/tutorials/marine_omics_bgc/tutorial.md %})
+---
+
+
+## Generate the RO-Crate
+
+Once you run the workflow you can generate the RO-Crate. To do so follow the [tutorial]({% link topics/fair/tutorials/ro-crate-in-galaxy/tutorial.md %})
+---
+
+
+### From workflow to RO-Crate
+
+
+---
+
+### The RO-Crate folder and architecture
+
+
+---
+
+### Let's dive into the ro-crate-metadata.json
+
+
+---
+
+### Description of the RO-Crate structure
+
+
+---
+
+### Galaxy Provenance
+
+
+---
+
+### Title and workflow details
+
+
+---
+
+### Dates
+
+
+---
+
+### Data
+
+
+---
+
+### First tool Prodigal
+
+
+---
+
+### Second tool Regex Find and Replace
+
+
+---
+
+### Third tool InterProScan
+
+
+---
+
+### Fourth tool SanntiS to build Genbank
+
+
+---
+
+### Fourth tool SanntiS to annotate biosynthetic gene clusters
+
+
+---
+
+### Details on the different files in the RO-Crate
+
+
+---
+
+### End of the ro-crate-metadata.json
+
+
+---
+
+### Thank you!
+
+Try to do the same on your workflow!
+
+
+
+
diff --git a/topics/galaxy-interface/metadata.yaml b/topics/galaxy-interface/metadata.yaml
index 5071040f57f69..b8a0078ee1126 100644
--- a/topics/galaxy-interface/metadata.yaml
+++ b/topics/galaxy-interface/metadata.yaml
@@ -5,6 +5,11 @@ title: "Using Galaxy and Managing your Data"
summary: "A collection of microtutorials explaining various features of the Galaxy user interface and manipulating data within Galaxy."
docker_image:
requirements:
+ -
+ type: "internal"
+ topic_name: introduction
+ tutorials:
+ - galaxy-intro-101
subtopics:
- id: upload
diff --git a/topics/galaxy-interface/tutorials/collections/faqs/index.md b/topics/galaxy-interface/tutorials/collections/faqs/index.md
index 9ce3fe4fce824..c66c9e96e0565 100644
--- a/topics/galaxy-interface/tutorials/collections/faqs/index.md
+++ b/topics/galaxy-interface/tutorials/collections/faqs/index.md
@@ -1,3 +1,5 @@
---
layout: faq-page
+redirect_from:
+- /topics/galaxy-interface/tutorials/processing-many-samples-at-once/faqs/index
---
diff --git a/topics/galaxy-interface/tutorials/processing-many-samples-at-once/faqs/visualisations_igv.md b/topics/galaxy-interface/tutorials/collections/faqs/visualisations_igv.md
similarity index 100%
rename from topics/galaxy-interface/tutorials/processing-many-samples-at-once/faqs/visualisations_igv.md
rename to topics/galaxy-interface/tutorials/collections/faqs/visualisations_igv.md
diff --git a/topics/galaxy-interface/tutorials/collections/tutorial.md b/topics/galaxy-interface/tutorials/collections/tutorial.md
index 552daa1e0b61e..aa8dab75e312f 100644
--- a/topics/galaxy-interface/tutorials/collections/tutorial.md
+++ b/topics/galaxy-interface/tutorials/collections/tutorial.md
@@ -2,6 +2,8 @@
layout: tutorial_hands_on
redirect_from:
- /topics/galaxy-data-manipulation/tutorials/collections/tutorial
+ - /topics/galaxy-data-manipulation/tutorials/processing-many-samples-at-once/tutorial
+ - /topics/galaxy-interface/tutorials/processing-many-samples-at-once/tutorial
title: "Using dataset collections"
zenodo_link: "https://doi.org/10.5281/zenodo.5119008"
@@ -46,6 +48,7 @@ recordings:
Here we will show Galaxy features designed to help with the analysis of large numbers of samples. When you have just a few samples - clicking through them is easy. But once you've got hundreds - it becomes very annoying. In Galaxy we have introduced **Dataset collections** that allow you to combine numerous datasets in a single entity that can be easily manipulated.
+
# Getting data
First, we need to upload datasets. Cut and paste the following URLs to Galaxy upload tool (see a {% icon tip %} **Tip** on how to do this [below](#tip-upload-fastqsanger-datasets-via-links)).
diff --git a/topics/galaxy-interface/tutorials/history-to-workflow/tutorial.md b/topics/galaxy-interface/tutorials/history-to-workflow/tutorial.md
index ea567c6ef4a3e..281d90bd57a44 100644
--- a/topics/galaxy-interface/tutorials/history-to-workflow/tutorial.md
+++ b/topics/galaxy-interface/tutorials/history-to-workflow/tutorial.md
@@ -54,7 +54,7 @@ This practical shows how to create a reusable analysis pipeline, called a *workf
> 1. An internet-connected computer. Galaxy can run on your laptop without an internet connection, but this practical requires access to resources on the web.
> 1. A web browser. [Firefox](https://www.mozilla.org/firefox) and [Google Chrome](https://www.google.com/chrome/) work well, as does [Safari](https://www.apple.com/safari/). Internet Explorer is known to have issues with Galaxy so avoid using that.
> 1. Access to a Galaxy instance, and an account on that instance. Galaxy is available in many ways. If you are doing this practical as part of a workshop, the instructor will tell you which instance to use. If you are doing this on your own, you can use [usegalaxy.org](https://usegalaxy.org).
-> 1. A Galaxy *history* from an analysis that you have already run. If you don't have one handy, you can use [this history](https://usegalaxy.org/u/tnabtaf/h/overlapping-genes-on-opposite-strands). on usegalaxy.org. Click **Import History** (top right) to bring that history into your workspace on usegalaxy.org. Any history will work with this tutorial, but that's the one used in the examples.
+> 1. A Galaxy *history* from an analysis that you have already run. If you don't have one handy, you can use [this history](https://usegalaxy.org/u/tnabtaf/h/overlapping-genes-on-opposite-strands-1). on usegalaxy.org. Click **Import History** (top right) to bring that history into your workspace on usegalaxy.org. Any history will work with this tutorial, but that's the one used in the examples.
{: .comment}
This tutorial is a good second step after running your first analysis on Galaxy.
diff --git a/topics/galaxy-interface/tutorials/processing-many-samples-at-once/tutorial.md b/topics/galaxy-interface/tutorials/processing-many-samples-at-once/tutorial.md
deleted file mode 100644
index 5c5d9c2d64a7f..0000000000000
--- a/topics/galaxy-interface/tutorials/processing-many-samples-at-once/tutorial.md
+++ /dev/null
@@ -1,184 +0,0 @@
----
-layout: tutorial_hands_on
-redirect_from:
- - /topics/galaxy-data-manipulation/tutorials/processing-many-samples-at-once/tutorial
-
-title: "Multisample Analysis"
-tags:
- - collections
-zenodo_link: ""
-level: Advanced
-questions:
-objectives:
-time_estimation: "1h"
-key_points:
-contributors:
- - nekrut
- - pajanne
-subtopic: manage
----
-
-Here we will show Galaxy features designed to help with the analysis of large numbers of samples. When you have just a few samples - clicking through them is easy. But once you've got hundreds - it becomes very annoying. In Galaxy we have introduced **Dataset collections** that allow you to combine numerous datasets in a single entity that can be easily manipulated.
-
-In this tutorial we assume the following:
-
-- you already have basic understanding of how Galaxy works
-- you have an account in Galaxy
-
->
->
-> In this tutorial, we will deal with:
->
-> 1. TOC
-> {:toc}
->
-{: .agenda}
-
-# Getting data
-[In this history](https://test.galaxyproject.org/u/anton/h/collections-1) are a few datasets we will be practicing with (as always with Galaxy tutorial you can upload your own data and play with it instead of the provided datasets):
-
-- `M117-bl_1` - family 117, mother, 1-st (**F**) read from **blood**
-- `M117-bl_2` - family 117, mother, 2-nd (**R**) read from **blood**
-- `M117-ch_1` - family 117, mother, 1-st (**F**) read from **cheek**
-- `M117-ch_1` - family 117, mother, 2-nd (**R**) read from **cheek**
-- `M117C1-bl_1`- family 117, child, 1-st (**F**) read from **blood**
-- `M117C1-bl_2`- family 117, child, 2-nd (**R**) read from **blood**
-- `M117C1-ch_1`- family 117, child, 1-st (**F**) read from **cheek**
-- `M117C1-ch_2`- family 117, child, 2-nd (**R**) read from **cheek**
-
-These datasets represent genomic DNA (enriched for mitochondria via a long range PCR) isolated from blood and cheek (buccal swab) of mother (`M117`) and her child (`M117C1`) that was sequenced on an Illumina miSeq machine as paired-read library (250-bp reads; see our [2014](http://www.pnas.org/content/111/43/15474.abstract) manuscript for **Methods**).
-
-# Creating a list of paired datasets
-
-If you imported [history]( https://test.galaxyproject.org/u/anton/h/collections-1) as described [above](https://github.com/nekrut/galaxy/wiki/Processing-many-samples-at-once#0-getting-data), your screen will look something like this:
-
-
-
-Now click the checkbox in  and you will see your history changing like this:
-
-
-
-Let's click `All`, which will select all datasets in the history, then click  and finally select **Build List of Dataset Pairs** from the following menu:
-
-
-
-The following wizard will appear:
-
-
-
-In this case Galaxy automatically assigned pairs using the `_1` and `_2` endings of dataset names. Let's however pretend that this did not happen. Click on **Unpair all** (highlighted in red in the figure above) link and then on **Clear** link (highlighted in blue in the figure above). The interface will change into its virgin state:
-
-
-
-Hopefully you remember that we have paired-end data in this scenario. Datasets containing the first (forward) and the second (reverse) read are differentiated by having `_1` and `_2` in the filename. We can use this feature in dataset collection wizard to pair our datasets. Type `_1` in the left **Filter this list** text box and `_2` in the right:
-
-
-
-You will see that the dataset collection wizard will automatically filter lists on each side of the interface:
-
-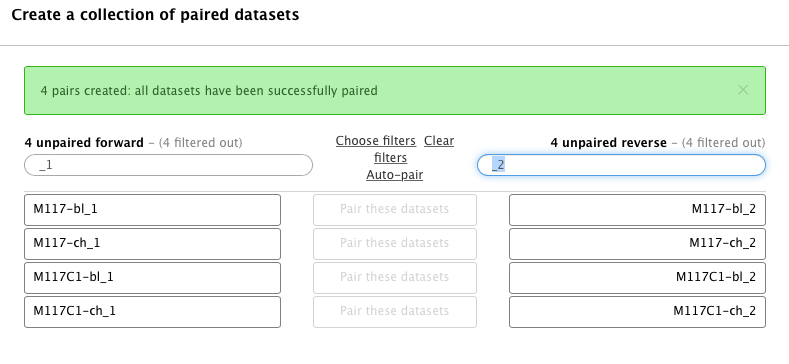
-
-Now you can either click **Auto pair** if pairs look good to you (proper combinations of datasets are listed in each line) or pair each forward/reverse group individually by pressing **Pair these datasets** button separating each pair:
-
-
-
-Now it is time to name the collection:
-
-
-
-and create the collection by clicking **Create list**. A new item will appear in the history as you can see on the panel **A** below. Clicking on collection will expand it to show four pairs it contains (panel **B**). Clicking individual pairs will expand them further to reveal **forward** and **reverse** datasets (panel **C**). Expanding these further will enable one to see individual datasets (panel **D**).
-
-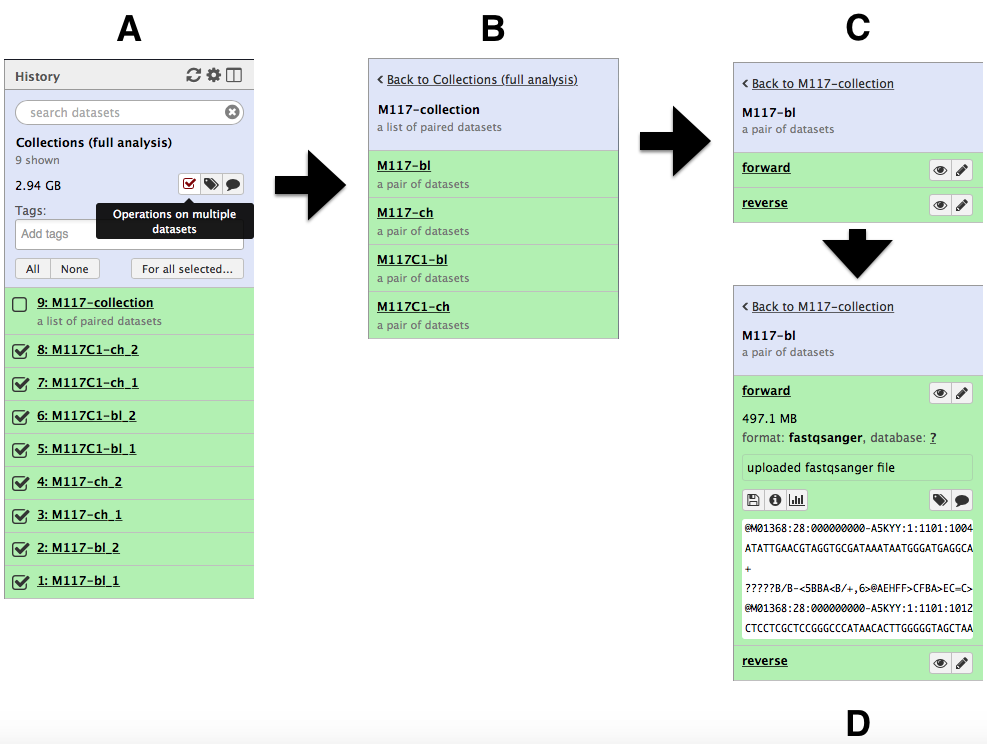
-
-# Using collections
-
-By now we see that a collection can be used to bundle a large number of items into a single history item. This means that many Galaxy tools will be able to process all datasets in a collection transparently to you. Let's try to map these datasets to human genome using `bwa-mem` mapper:
-
-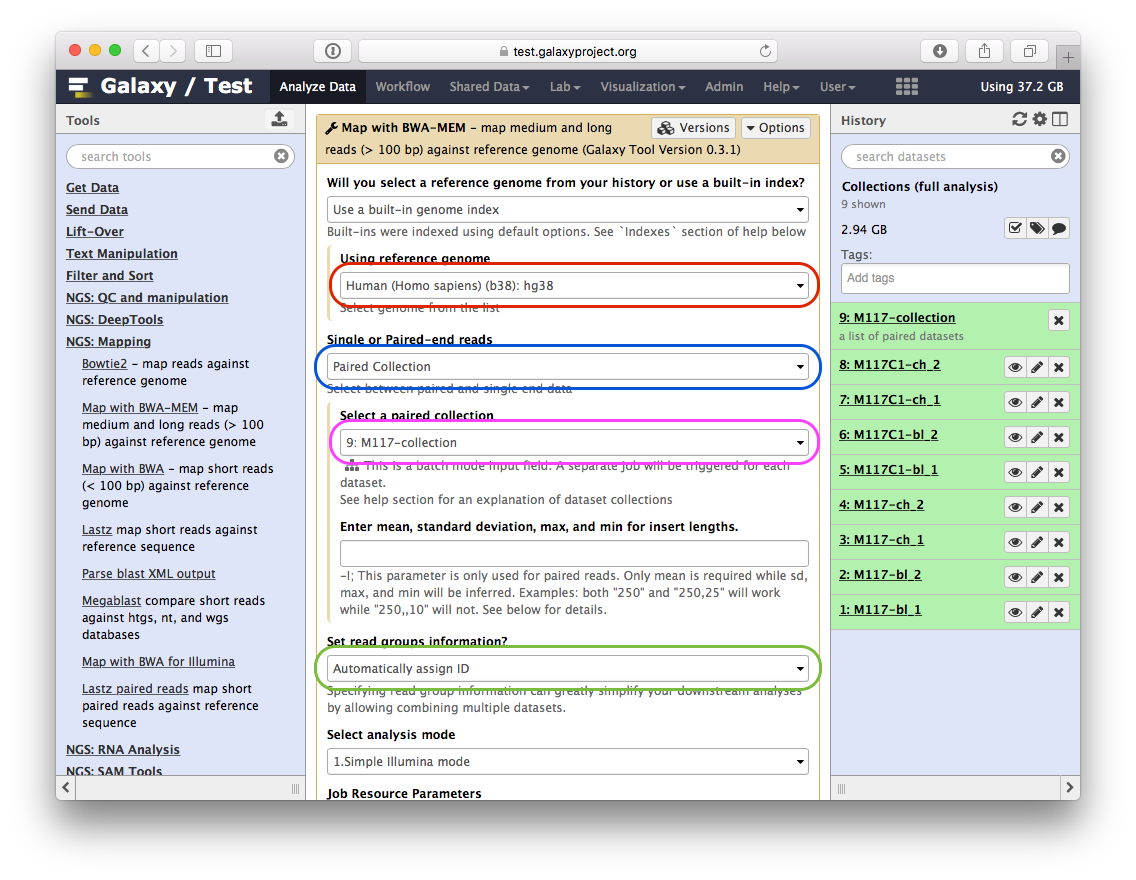
-
-Here is what you need to do:
-
-- set **Using reference genome** to `hg38` (red outline);
-- set **Single or Paired-end reads** to `Paired collection` (blue outline);
-- select `M177-collection` from **Select a paired collection** dropdown (magenta outline);
-- In **Set read groups information** select `Automatically assign ID` (green outline);
-- scroll down and click **Execute**.
-
-You will see jobs being submitted and new datasets appearing in the history. IN particular below you can see that Galaxy has started four jobs (two yellow and two gray). This is because we have eight paired datasets with each pair being processed separately by `bwa-mem`. As a result we have four `bwa-mem` runs:
-
-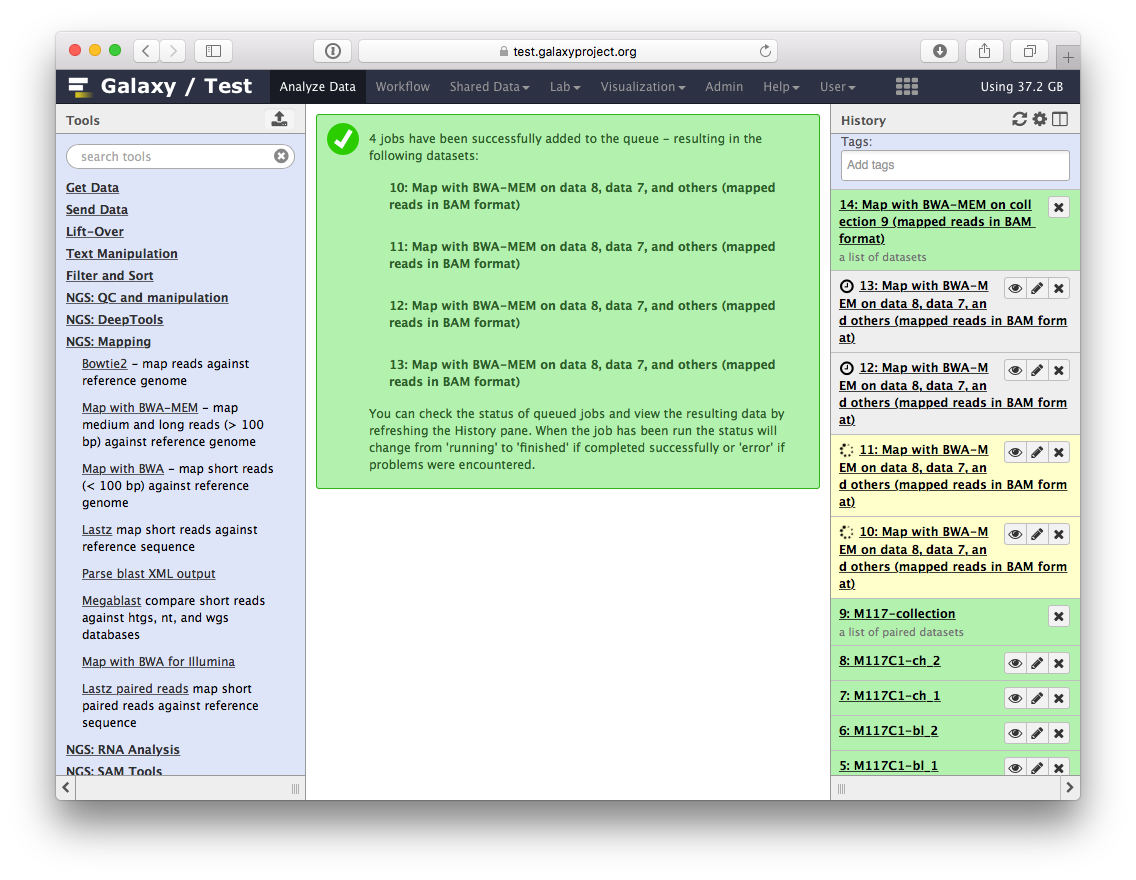
-
-Once these jobs are finished they will disappear from the history and all results will be represented as a new collection:
-
-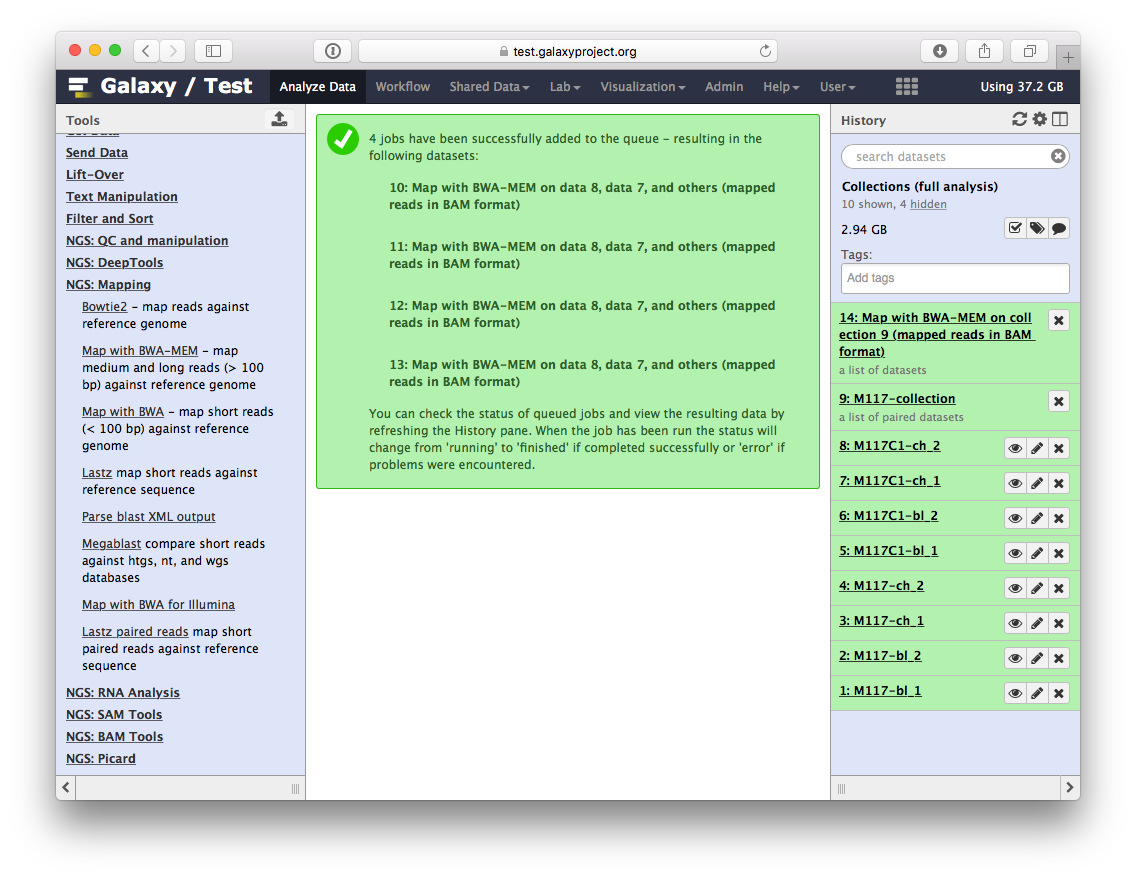
-
-Let's look at this collection by clicking on it (panel **A** in the figure below). You can see that now this collection is no longer paired (compared to the collection we created in the beginning of this tutorial). This is because `bwa-mem` takes forward and reverse data as input, but produces only a single BAM dataset as the output. So what we have in the result is a *list* of four dataset (BAM files; panels **B** and **C**).
-
-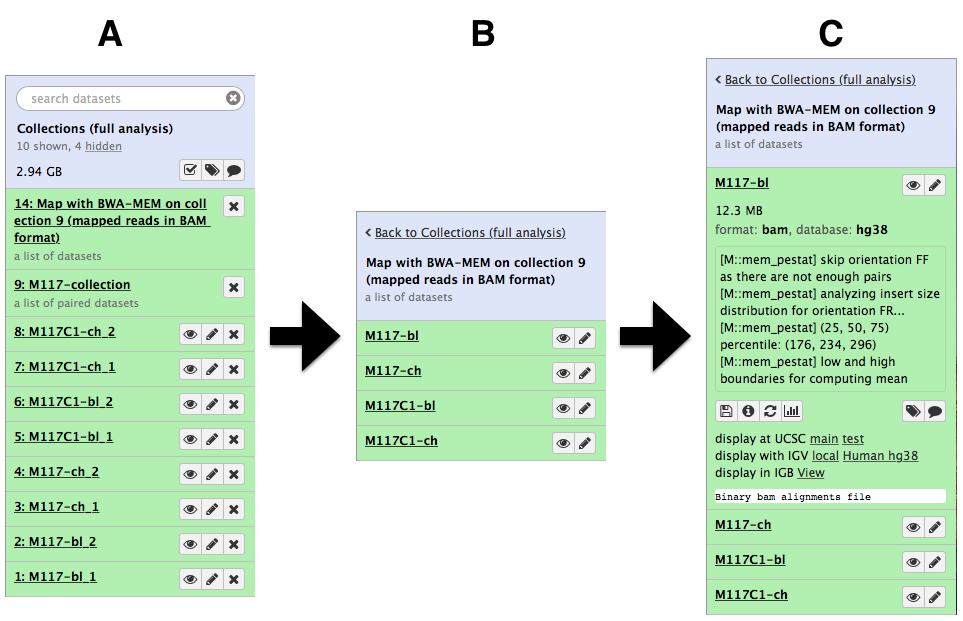
-
-# Processing collection as a single entity
-
-Now that `bwa-mem` has finished and generated a collection of BAM datasets we can continue to analyze the entire collection as a single Galaxy '*item*'.
-
-## Ensuring consistency of BAM dataset
-
-Let's perform cleanup of our BAM files with `cleanSam` utility from the **Picard** package:
-
-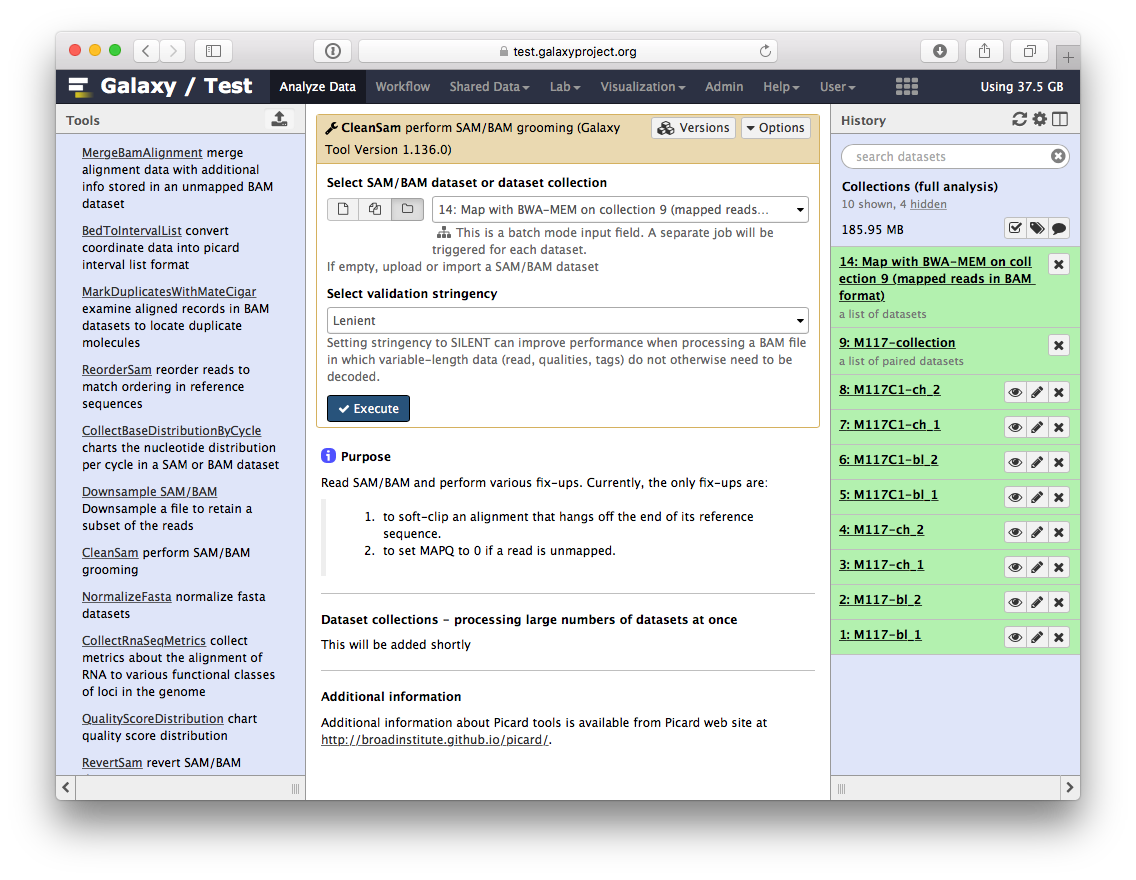
-
-If you look at the picture above carefully, you will see that the **Select SAM/BAM dataset or dataset collection** parameter is empty (it says `No sam or bam datasets available.`). This is because we do not have single SAM or BAM datasets in the history. Instead we have a collection. So all you need to do is to click on the **folder** () button and you will our BAM collection selected:
-
-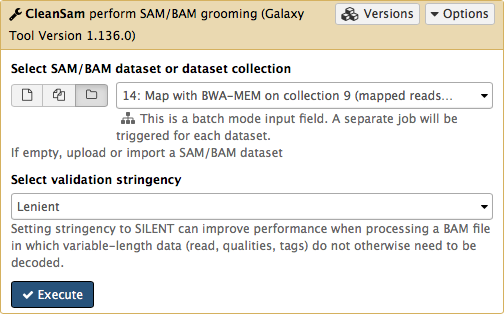
-
-Click **Run Tool**. As an output this tool will produce a collection contained cleaned data.
-
-## Retaining 'proper pairs'
-
-Now let's clean the dataset further by only preserving truly paired reads (reads satisfying two requirements: (1) read is paired, and (2) it is mapped as a proper pair). For this we will use `Filter SAM or BAM` tools from **SAMTools** collection:
-
-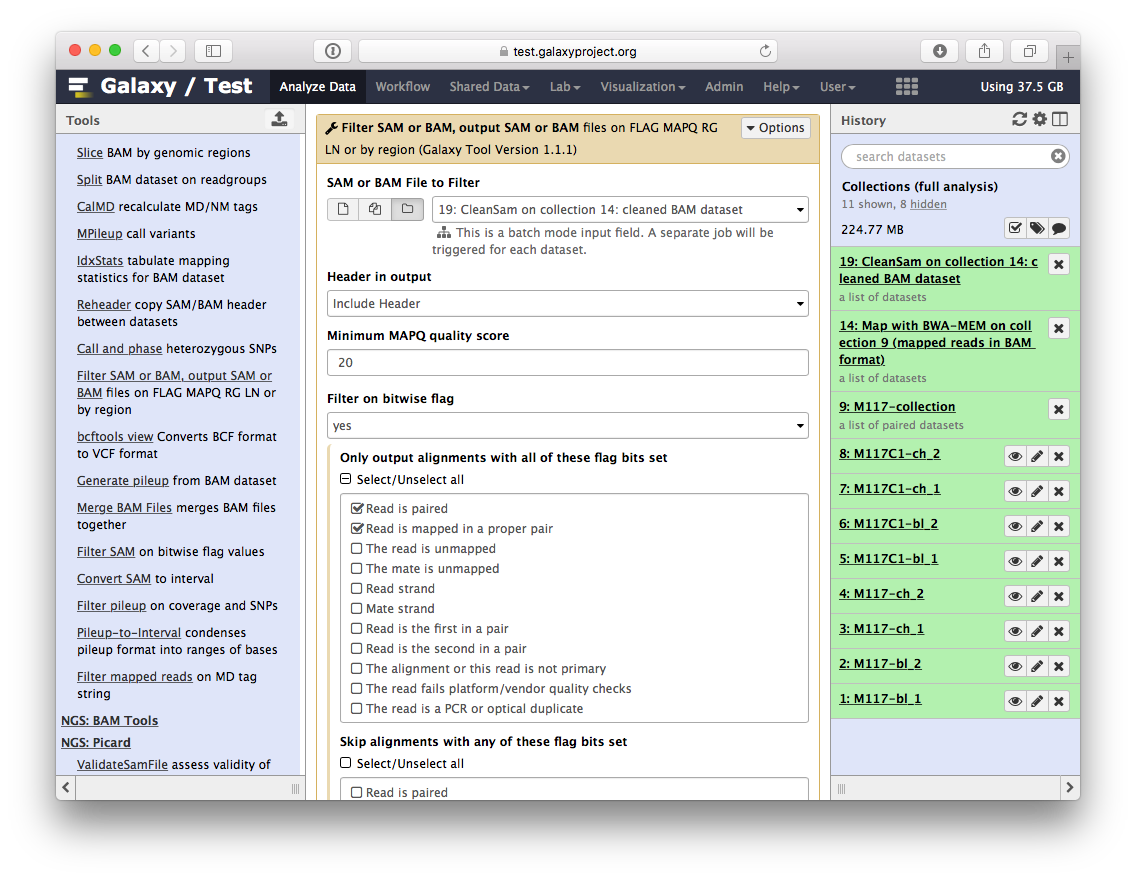
-
-parameters should be set as shown below. By setting mapping quality to `20` we avoid reads mapping to multiple locations and by using **Filter on bitwise flag** option we ensure that the resulting dataset will contain only properly paired reads. This operation will produce yet another collection containing now filtered datasets.
-
-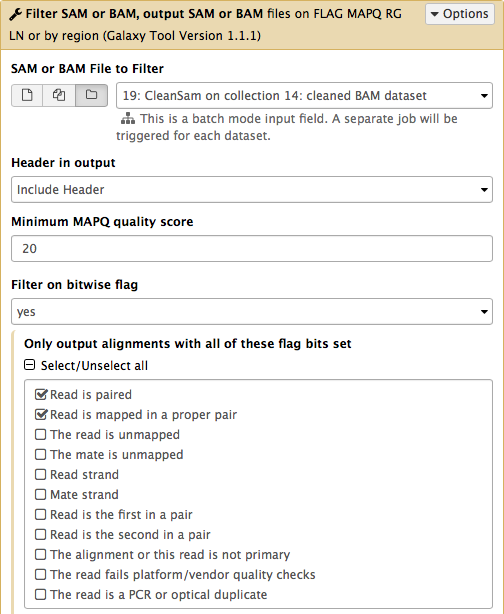
-
-## Merging collection into a single dataset
-
-The beauty of BAM datasets is that they can be combined in a single entity using so called *Read group* ([learn more](https://wiki.galaxyproject.org/Learn/GalaxyNGS101#Understanding_and_manipulating_SAM.2FBAM_datasets) about Read Groups on old wiki, which will be migrated here shortly). This allows to bundle reads from multiple experiments into a single dataset where read identity is maintained by labelling every sequence with *read group* tags. So let's finally reduce this collection to a single BAM dataset. For this we will use `MergeSamFiles` tool for the `Picard` suite:
-
-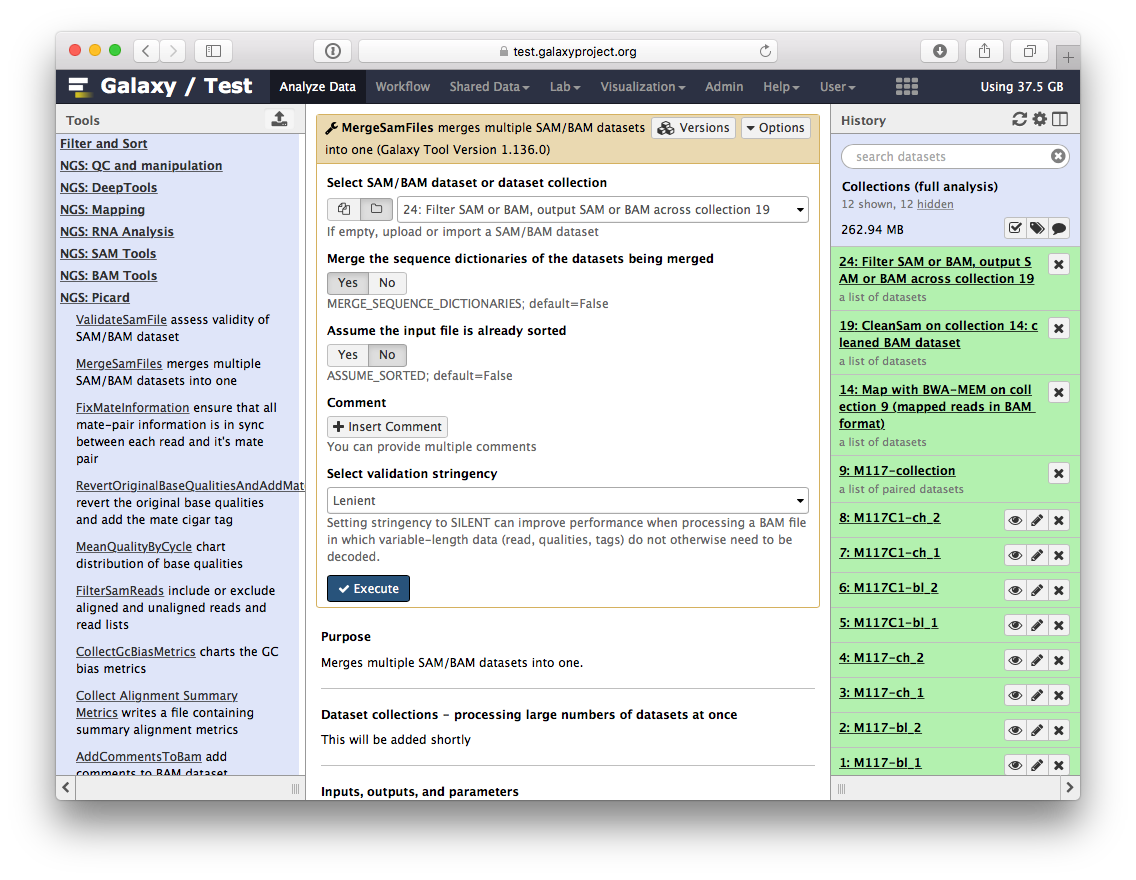
-
-Here we select the collection generated by the filtering tool described above in [3.1](https://github.com/nekrut/galaxy/wiki/Processing-many-samples-at-once#31-retaining-proper-pairs):
-
-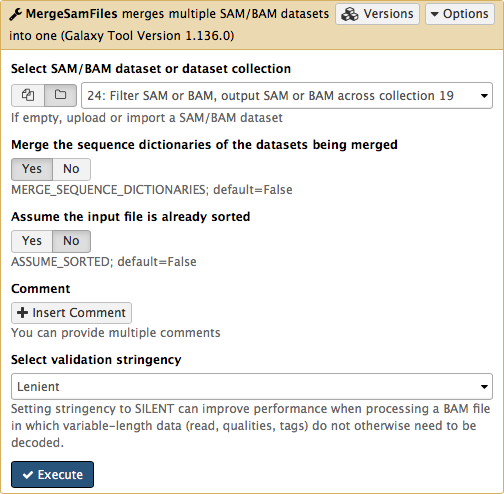
-
-This operation will **not** generate a collection. Instead, it will generate a single BAM dataset containing mapped reads from our four samples (`M117-bl`, `M117-ch`, `M117C1-bl`, and `M117C1-ch`).
-
-# Let's look at what we've got!
-
-So we have one BAM dataset combining everything we've done so far. Let's look at the contents of this dataset using a genome browser. First, we will need to downsample the dataset to avoiding overwhelming the browser. For this we will use `Downsample SAM/BAM` tool:
-
-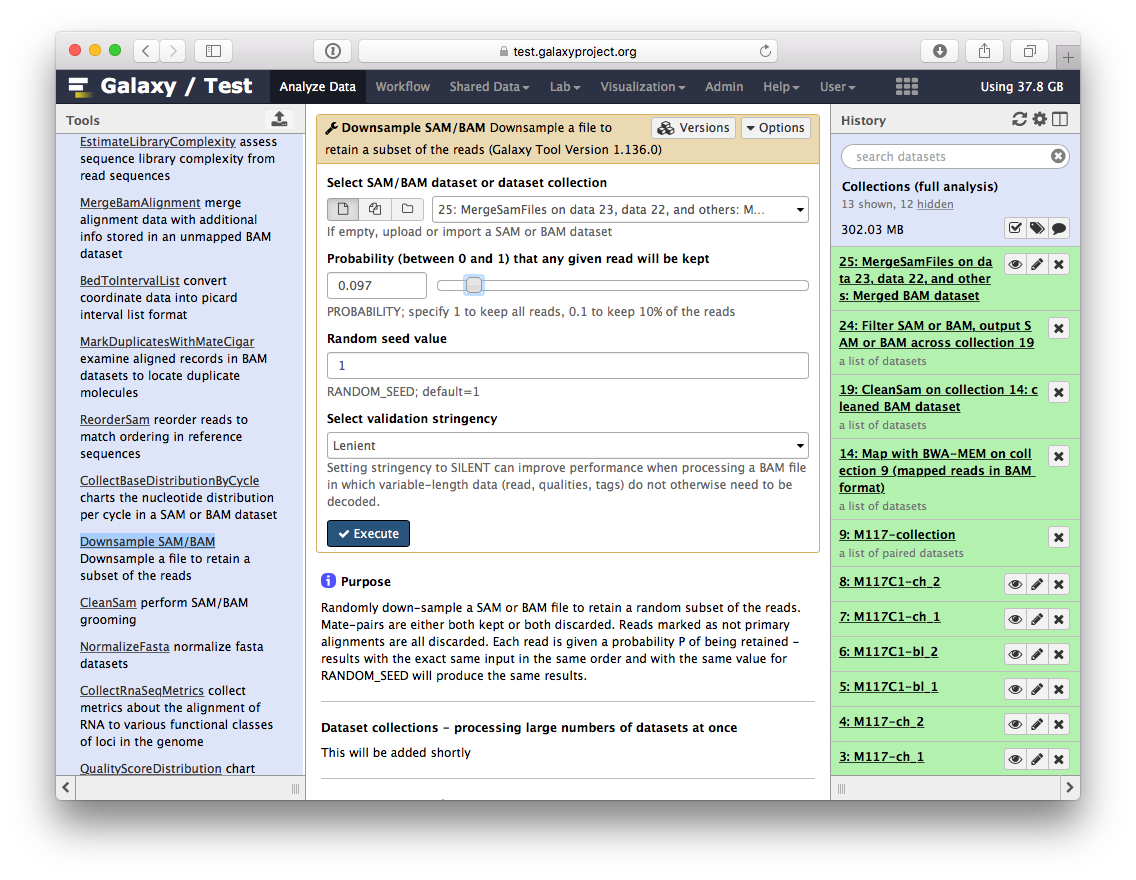
-
-Set **Probability (between 0 and 1) that any given read will be kept** to roughly `5%` (or `0.05`) using the slider control:
-
-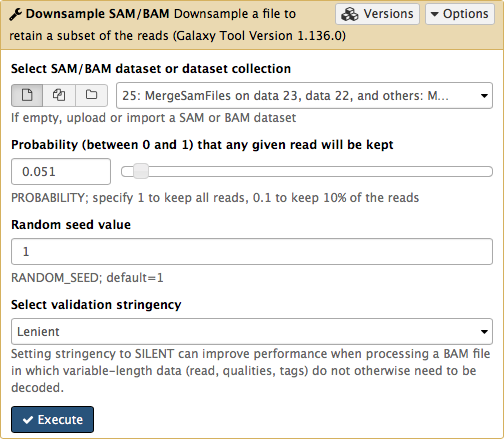
-
-This will generate another BAM dataset containing only 5% of the original reads and much smaller as a result. Click on this dataset and you will see links to various genome browsers:
-
-
-
-Click the **Human hg38** link in the **display with IGV** line as highlighted above ([learn](https://wiki.galaxyproject.org/Learn/GalaxyNGS101#Visualizing_multiple_datasets_in_Integrated_Genome_Viewer_.28IGV.29) more about displaying Galaxy data in IGV with this [movie](https://vimeo.com/123442619#t=4m16s)). Below is an example generated with IGV on these data. In this screenshot reads are colored by read group (four distinct colors). A yellow inset displays additional information about a single read. One can see that this read corresponds to read group `M117-bl`.
-
-
-
-# We did not fake this:
-The two histories and the workflow described in this page are accessible directly from this page below:
-
-* History [**Collections**]( https://test.galaxyproject.org/u/anton/h/collections-1)
-* History [**Collections (full analysis)**]( https://test.galaxyproject.org/u/anton/h/collections-full-analysis)
-
-From there you can import histories to make them your own.
-
-# If things don't work...
-...you need to complain. Use [Galaxy's Help Channel](https://help.galaxyproject.org/) to do this.
diff --git a/topics/genome-annotation/tutorials/helixer/tutorial.md b/topics/genome-annotation/tutorials/helixer/tutorial.md
index d99a9ecde5f3d..7f732f11e125f 100644
--- a/topics/genome-annotation/tutorials/helixer/tutorial.md
+++ b/topics/genome-annotation/tutorials/helixer/tutorial.md
@@ -224,6 +224,35 @@ This gives information about the completeness of the Helixer annotation. A good
>
{: .comment}
+## Evaluation with **OMArk**
+
+[OMArk](https://github.com/DessimozLab/OMArk) is proteome quality assessment software. It provides measures of proteome completeness, characterises the consistency of all protein-coding genes with their homologues and identifies the presence of contamination by other species. OMArk is based on the OMA orthology database, from which it exploits orthology relationships, and on the OMAmer software for rapid placement of all proteins in gene families.
+
+OMArk's analysis is based on HOGs (Hierarchical Orthologous Groups), which play a central role in its assessment of the completeness and coherence of gene sets. HOGs make it possible to compare the genes of a given species with groups of orthologous genes conserved across a taxonomic clade.
+
+> OMArk on extracted protein sequences
+>
+> 1. {% tool [OMArk](toolshed.g2.bx.psu.edu/repos/iuc/omark/omark/0.3.0+galaxy2) %} with the following parameters:
+> - {% icon param-file %} *"Protein sequences"*: `gffread: pep.fa`
+> - *"OMAmer database*: select `LUCA-v2.0.0`
+> - In *"Which outputs should be generated"*: select `Detailed summary`
+>
+{: .hands_on}
+
+The OMArk tool generated an output file in .txt format containing detailed information on the assessment of the completeness, consistency and species composition of the proteome analysed. This report includes statistics on conserved genes, the proportion of duplications, missing genes and the identification of reference lineages.
+
+> What can we deduce from these results?
+>
+> - Number of conserved HOGs: OMArk has identified a set of 5622 HOGs which are thought to be conserved in the majority of species in the Mucorineae clade.
+> - 85.52% of genes are complete, so the annotation is of good quality in terms of genomic completeness.
+> - Number of proteins in the whole proteome: 19 299. Of which 62.83% are present and 30.94% of the proteome does not share sufficient similarities with known gene families.
+> - No contamination detected.
+> - The OMArk analysis is based on the Mucorineae lineage, a more recent and specific clade than that used in the BUSCO assessment, which selected the Mucorales as the reference group.
+{: .comment}
+
+
+
+
# Visualisation with a genome browser
You can visualize the annotation generated using a genomic browser like [JBrowse](https://jbrowse.org/jbrowse1.html). This browser enables you to navigate along the chromosomes of the genome and view the structure of each predicted gene.
diff --git a/topics/genome-annotation/tutorials/helixer/workflows/Helixer-tests.yml b/topics/genome-annotation/tutorials/helixer/workflows/Helixer-tests.yml
index 387276199fc22..94ba84e720730 100644
--- a/topics/genome-annotation/tutorials/helixer/workflows/Helixer-tests.yml
+++ b/topics/genome-annotation/tutorials/helixer/workflows/Helixer-tests.yml
@@ -1,34 +1,73 @@
-- doc: Test outline for Helixer
+- doc: Test outline for Helixer Workflow
job:
- Genome:
+ Input:
class: File
+ location: https://zenodo.org/records/13890774/files/genome_masked.fa?download=1
filetype: fasta
- path: 'test-data/sequence.fasta'
outputs:
- Helixer:
- asserts:
- has_text:
- text: '##gff-version 3'
- compleasm full table:
- asserts:
- has_text:
- text: '10251at4827'
- compleasm full table busco:
- asserts:
- has_text:
- text: '10251at4827'
- compleasm miniprot:
- asserts:
- has_text:
- text: '10011at4827'
- compleasm translated protein:
- asserts:
- has_text:
- text: 'CCTCCCTCCCTCCCTCLINRLYRERLFFLGQEVDTEISNQLISLMIYLSIEKDTKDLYLFINSPGGWVISGMAIYDTMQFVRPDVQTICMGLAASIASFILVGGAITKRIAFPHAWVMIHQPASSFYEAQTGEFILEAEELLKLRETITRVYVQRTGKPIWVVSEDMERDVFMSATEAQAHGIVDLVACCTCCCTCC'
-
- BUSCO sum:
- asserts:
- has_text:
- text: '2449'
- text: '5.5.0'
+ helixer_output:
+ location: https://zenodo.org/records/13890774/files/Helixer.gff3?download=1
+ compare: sim_size
+ delta: 300000
+
+ busco_sum_geno:
+ location: https://zenodo.org/records/13890774/files/Busco_short_summary_genome.txt?download=1
+ compare: sim_size
+ delta: 30000
+ busco_gff_geno:
+ location: https://zenodo.org/records/13890774/files/Busco_GFF_genome.gff3?download=1
+ compare: sim_size
+ delta: 30000
+ summary_image_geno:
+ location: https://zenodo.org/records/13890774/files/Busco_summary_image_genome.png?download=1
+ compare: sim_size
+ delta: 30000
+ busco_missing_geno:
+ location: https://zenodo.org/records/13890774/files/Busco_missing_buscos_genome.tabular?download=1
+ compare: sim_size
+ delta: 30000
+ busco_table_geno:
+ location: https://zenodo.org/records/13890774/files/Busco_full_table_genome.tabular?download=1
+ compare: sim_size
+ delta: 30000
+
+ gffread_pep:
+ location: https://zenodo.org/records/13890774/files/gffread_pep.fasta?download=1
+ compare: sim_size
+ delta: 30000
+
+ summary:
+ location: https://zenodo.org/records/13890774/files/genome_annotation_statistics_summary.txt?download=1
+ compare: sim_size
+ delta: 30000
+ graphs:
+ location: https://zenodo.org/records/13890774/files/genome_annotation_statistics_graphs.pdf?download=1
+ compare: sim_size
+ delta: 30000
+
+ summary_image_pep:
+ location: https://zenodo.org/records/13902305/files/Busco_pep_summary_image.png?download=1
+ compare: sim_size
+ delta: 30000
+ busco_table_pep:
+ location: https://zenodo.org/records/13890774/files/Busco_full_table_pep.tabular?download=1
+ compare: sim_size
+ delta: 30000
+ busco_sum_pep:
+ location: https://zenodo.org/records/13890774/files/Busco_short_summary_pep.txt?download=1
+ compare: sim_size
+ delta: 30000
+ busco_gff_pep:
+ location: https://zenodo.org/records/13890774/files/Busco_GFF_pep.gff3?download=1
+ compare: sim_size
+ delta: 30000
+ busco_missing_pep:
+ location: https://zenodo.org/records/13890774/files/Busco_missing_buscos_pep.tabular?download=1
+ compare: sim_size
+ delta: 30000
+
+ omark_detail_sum:
+ location: https://zenodo.org/records/13890774/files/OMArk_Detailed_summary.txt?download=1
+ compare: sim_size
+ delta: 30000
diff --git a/topics/genome-annotation/tutorials/helixer/workflows/Helixer.ga b/topics/genome-annotation/tutorials/helixer/workflows/Helixer.ga
index 42bfae7e428ec..32bc124526514 100644
--- a/topics/genome-annotation/tutorials/helixer/workflows/Helixer.ga
+++ b/topics/genome-annotation/tutorials/helixer/workflows/Helixer.ga
@@ -1,6 +1,105 @@
{
"a_galaxy_workflow": "true",
- "annotation": "Structural genome annotation with Helixer",
+ "annotation": "This workflow allows you to annotate a genome with Helixer and evaluate the quality of the annotation using BUSCO and Genome Annotation statistics. GFFRead is also used to predict protein sequences derived from this annotation, and BUSCO and OMArk are used to assess proteome quality. ",
+ "comments": [
+ {
+ "child_steps": [
+ 4,
+ 2
+ ],
+ "color": "lime",
+ "data": {
+ "title": "Evaluation - Genome annotation"
+ },
+ "id": 2,
+ "position": [
+ 468.3,
+ 902.5
+ ],
+ "size": [
+ 496.5,
+ 356.1
+ ],
+ "type": "frame"
+ },
+ {
+ "child_steps": [
+ 3
+ ],
+ "color": "orange",
+ "data": {
+ "title": "Protein prediction with Helixer annotation"
+ },
+ "id": 1,
+ "position": [
+ 628.9,
+ 255.2
+ ],
+ "size": [
+ 258,
+ 275
+ ],
+ "type": "frame"
+ },
+ {
+ "child_steps": [
+ 1
+ ],
+ "color": "blue",
+ "data": {
+ "title": "Annotation step"
+ },
+ "id": 0,
+ "position": [
+ 238.5,
+ 458.79999999999995
+ ],
+ "size": [
+ 240,
+ 183
+ ],
+ "type": "frame"
+ },
+ {
+ "child_steps": [
+ 5
+ ],
+ "color": "pink",
+ "data": {
+ "title": "Visualization"
+ },
+ "id": 4,
+ "position": [
+ 1045.3,
+ 680.0
+ ],
+ "size": [
+ 240,
+ 244.5
+ ],
+ "type": "frame"
+ },
+ {
+ "child_steps": [
+ 6,
+ 7
+ ],
+ "color": "turquoise",
+ "data": {
+ "title": "Evaluation - Predicted protein from annotation"
+ },
+ "id": 3,
+ "position": [
+ 1104.4,
+ 0.0
+ ],
+ "size": [
+ 312,
+ 563
+ ],
+ "type": "frame"
+ }
+ ],
"creator": [
{
"class": "Person",
@@ -10,37 +109,40 @@
],
"format-version": "0.1",
"license": "MIT",
- "name": "Training - Helixer",
+ "name": "annotation_helixer",
+ "report": {
+ "markdown": "\n# Workflow Execution Report\n\n## Workflow Inputs\n```galaxy\ninvocation_inputs()\n```\n\n## Workflow Outputs\n```galaxy\ninvocation_outputs()\n```\n\n## Workflow\n```galaxy\nworkflow_display()\n```\n"
+ },
"steps": {
"0": {
- "annotation": "Genome input (fasta format)",
+ "annotation": "Input dataset containing genomic sequences in FASTA format",
"content_id": null,
"errors": null,
"id": 0,
"input_connections": {},
"inputs": [
{
- "description": "Genome input (fasta format)",
- "name": "Genome"
+ "description": "Input dataset containing genomic sequences in FASTA format",
+ "name": "Input"
}
],
- "label": "Genome",
+ "label": "Input",
"name": "Input dataset",
"outputs": [],
"position": {
- "left": 0.0,
- "top": 393.5426496082689
+ "left": 0,
+ "top": 812.5362146249206
},
"tool_id": null,
- "tool_state": "{\"optional\": false, \"tag\": null}",
+ "tool_state": "{\"optional\": false, \"format\": [\"fasta\"], \"tag\": \"\"}",
"tool_version": null,
"type": "data_input",
- "uuid": "332fb9cd-3819-4e81-8aa5-c6cc6fbda4f7",
+ "uuid": "e267e1df-03ae-4b70-98ce-65ea177a172e",
"when": null,
"workflow_outputs": []
},
"1": {
- "annotation": "Structural annotation step",
+ "annotation": "Helixer tool for genomic annotation",
"content_id": "toolshed.g2.bx.psu.edu/repos/genouest/helixer/helixer/0.3.3+galaxy1",
"errors": null,
"id": 1,
@@ -53,7 +155,7 @@
"inputs": [
{
"description": "runtime parameter for tool Helixer",
- "name": "input"
+ "name": "input_model"
}
],
"label": "Helixer",
@@ -65,33 +167,33 @@
}
],
"position": {
- "left": 495.6559490752344,
- "top": 126.52477912175132
+ "left": 258.5333251953125,
+ "top": 498.8339807128906
},
"post_job_actions": {},
"tool_id": "toolshed.g2.bx.psu.edu/repos/genouest/helixer/helixer/0.3.3+galaxy1",
"tool_shed_repository": {
- "changeset_revision": "e3846dc36c4d",
+ "changeset_revision": "c2fc4ac35199",
"name": "helixer",
"owner": "genouest",
"tool_shed": "toolshed.g2.bx.psu.edu"
},
- "tool_state": "{\"input\": {\"__class__\": \"RuntimeValue\"}, \"lineages\": \"fungi\", \"option_overlap\": {\"use_overlap\": \"true\", \"__current_case__\": 0, \"overlap_offset\": null, \"overlap_core_length\": null}, \"post_processing\": {\"window_size\": \"100\", \"edge_threshold\": \"0.1\", \"peak_threshold\": \"0.8\", \"min_coding_length\": \"100\"}, \"size\": \"8\", \"species\": null, \"subsequence_length\": null, \"__page__\": null, \"__rerun_remap_job_id__\": null}",
+ "tool_state": "{\"input\": {\"__class__\": \"ConnectedValue\"}, \"input_model\": {\"__class__\": \"RuntimeValue\"}, \"lineages\": \"land_plant\", \"option_overlap\": {\"use_overlap\": \"true\", \"__current_case__\": 0, \"overlap_offset\": null, \"overlap_core_length\": null}, \"post_processing\": {\"window_size\": \"100\", \"edge_threshold\": \"0.1\", \"peak_threshold\": \"0.8\", \"min_coding_length\": \"100\"}, \"size\": \"8\", \"species\": null, \"subsequence_length\": null, \"__page__\": null, \"__rerun_remap_job_id__\": null}",
"tool_version": "0.3.3+galaxy1",
"type": "tool",
- "uuid": "c151bbb2-deb9-4fb6-a047-7a92ede3171a",
+ "uuid": "f60cf54d-31f2-4395-bb55-4916828cd211",
"when": null,
"workflow_outputs": [
{
- "label": "Helixer",
+ "label": "helixer_output",
"output_name": "output",
- "uuid": "9ebeb90d-1528-494c-a88d-50f28836d7c7"
+ "uuid": "fe43bcd6-5f99-4fd3-b184-2d6bfb340030"
}
]
},
"2": {
- "annotation": "Compleam is described as a faster and more accurate reimplementation of Busco.",
- "content_id": "toolshed.g2.bx.psu.edu/repos/iuc/compleasm/compleasm/0.2.5+galaxy0",
+ "annotation": "Completeness assessment of the genome using the Busco tool",
+ "content_id": "toolshed.g2.bx.psu.edu/repos/iuc/busco/busco/5.7.1+galaxy0",
"errors": null,
"id": 2,
"input_connections": {
@@ -101,163 +203,135 @@
}
},
"inputs": [],
- "label": "compleasm",
- "name": "compleasm",
+ "label": "Busco on genome",
+ "name": "Busco",
"outputs": [
{
- "name": "full_table_busco",
- "type": "tsv"
+ "name": "busco_sum",
+ "type": "txt"
},
{
- "name": "full_table",
- "type": "tsv"
+ "name": "busco_table",
+ "type": "tabular"
},
{
- "name": "miniprot",
- "type": "gff3"
+ "name": "busco_missing",
+ "type": "tabular"
},
{
- "name": "translated_protein",
- "type": "fasta"
+ "name": "summary_image",
+ "type": "png"
+ },
+ {
+ "name": "busco_gff",
+ "type": "gff3"
}
],
"position": {
- "left": 677.3732598347183,
- "top": 639.0246967831238
+ "left": 744.7633406324078,
+ "top": 942.4706486763349
},
"post_job_actions": {},
- "tool_id": "toolshed.g2.bx.psu.edu/repos/iuc/compleasm/compleasm/0.2.5+galaxy0",
+ "tool_id": "toolshed.g2.bx.psu.edu/repos/iuc/busco/busco/5.7.1+galaxy0",
"tool_shed_repository": {
- "changeset_revision": "47f9f4d13d2c",
- "name": "compleasm",
+ "changeset_revision": "2babe6d5c561",
+ "name": "busco",
"owner": "iuc",
"tool_shed": "toolshed.g2.bx.psu.edu"
},
- "tool_state": "{\"__input_ext\": \"input\", \"busco_database\": \"v5\", \"chromInfo\": \"/shared/ifbstor1/galaxy/mutable-config/tool-data/shared/ucsc/chrom/?.len\", \"input\": {\"__class__\": \"ConnectedValue\"}, \"lineage_dataset\": \"mucorales_odb10\", \"mode\": \"busco\", \"outputs\": [\"full_table_busco\", \"full_table\", \"miniprot\", \"translated_protein\"], \"specified_contigs\": null, \"__page__\": null, \"__rerun_remap_job_id__\": null}",
- "tool_version": "0.2.5+galaxy0",
+ "tool_state": "{\"adv\": {\"evalue\": \"0.001\", \"limit\": \"3\", \"contig_break\": \"10\"}, \"busco_mode\": {\"mode\": \"geno\", \"__current_case__\": 0, \"use_augustus\": {\"use_augustus_selector\": \"augustus\", \"__current_case__\": 2, \"aug_prediction\": {\"augustus_mode\": \"no\", \"__current_case__\": 0}, \"long\": false}}, \"input\": {\"__class__\": \"ConnectedValue\"}, \"lineage\": {\"lineage_mode\": \"auto_detect\", \"__current_case__\": 0, \"auto_lineage\": \"--auto-lineage\"}, \"lineage_conditional\": {\"selector\": \"cached\", \"__current_case__\": 0, \"cached_db\": \"v5\"}, \"outputs\": [\"short_summary\", \"image\", \"gff\", \"missing\"], \"__page__\": null, \"__rerun_remap_job_id__\": null}",
+ "tool_version": "5.7.1+galaxy0",
"type": "tool",
- "uuid": "e95adff9-488d-41af-b55c-c137ab9667cf",
+ "uuid": "c0e4cca7-0bc3-4ef2-81b2-c990b1b77d87",
"when": null,
"workflow_outputs": [
{
- "label": "compleasm miniprot",
- "output_name": "miniprot",
- "uuid": "e0d3aa0b-d18b-41af-a6fc-28a8688fe9a5"
+ "label": "busco_missing_geno",
+ "output_name": "busco_missing",
+ "uuid": "d039ef78-640f-4f7d-b449-69fac1a25130"
},
{
- "label": "compleasm full table",
- "output_name": "full_table",
- "uuid": "93b908a5-e39f-4cfb-84c3-edf4ab8b71cf"
+ "label": "busco_gff_geno",
+ "output_name": "busco_gff",
+ "uuid": "961890cc-7a33-422a-ab09-b787e3592dd1"
},
{
- "label": "compleasm full table busco",
- "output_name": "full_table_busco",
- "uuid": "7c9cb7e2-dfac-4aa1-b87d-f6a2eb7e21a5"
+ "label": "busco_sum_geno",
+ "output_name": "busco_sum",
+ "uuid": "bf09f09a-b403-4517-9a1a-acece8f36735"
},
{
- "label": "compleasm translated protein",
- "output_name": "translated_protein",
- "uuid": "9215afe6-4aad-4d43-a3a1-c2c5ab0ea1e2"
+ "label": "summary_image_geno",
+ "output_name": "summary_image",
+ "uuid": "3232c386-3c31-4989-ac76-02722ea2d79b"
+ },
+ {
+ "label": "busco_table_geno",
+ "output_name": "busco_table",
+ "uuid": "5cbbd77a-f521-4ee6-b990-a494b7671534"
}
]
},
"3": {
- "annotation": "BSUCO assesses the quality of genomic data. The tool inspects the transcription sequences of predicted genes.",
- "content_id": "toolshed.g2.bx.psu.edu/repos/iuc/busco/busco/5.5.0+galaxy0",
+ "annotation": "Converts GFF files to other formats, such as FASTA",
+ "content_id": "toolshed.g2.bx.psu.edu/repos/devteam/gffread/gffread/2.2.1.4+galaxy0",
"errors": null,
"id": 3,
"input_connections": {
"input": {
+ "id": 1,
+ "output_name": "output"
+ },
+ "reference_genome|genome_fasta": {
"id": 0,
"output_name": "output"
}
},
"inputs": [
{
- "description": "runtime parameter for tool Busco",
- "name": "input"
+ "description": "runtime parameter for tool gffread",
+ "name": "chr_replace"
+ },
+ {
+ "description": "runtime parameter for tool gffread",
+ "name": "reference_genome"
}
],
- "label": "BUSCO",
- "name": "Busco",
+ "label": "Gffread",
+ "name": "gffread",
"outputs": [
{
- "name": "busco_sum",
- "type": "txt"
- },
- {
- "name": "busco_table",
- "type": "tabular"
- },
- {
- "name": "busco_missing",
- "type": "tabular"
- },
- {
- "name": "summary_image",
- "type": "png"
- },
- {
- "name": "busco_gff",
- "type": "gff3"
- },
- {
- "name": "busco_miniprot",
- "type": "gff3"
+ "name": "output_pep",
+ "type": "fasta"
}
],
"position": {
- "left": 948.5586447752132,
- "top": 415.14516016871283
+ "left": 658.9081573207637,
+ "top": 316.7812237670679
},
"post_job_actions": {},
- "tool_id": "toolshed.g2.bx.psu.edu/repos/iuc/busco/busco/5.5.0+galaxy0",
+ "tool_id": "toolshed.g2.bx.psu.edu/repos/devteam/gffread/gffread/2.2.1.4+galaxy0",
"tool_shed_repository": {
- "changeset_revision": "ea8146ee148f",
- "name": "busco",
- "owner": "iuc",
+ "changeset_revision": "3e436657dcd0",
+ "name": "gffread",
+ "owner": "devteam",
"tool_shed": "toolshed.g2.bx.psu.edu"
},
- "tool_state": "{\"adv\": {\"evalue\": \"0.001\", \"limit\": \"3\", \"contig_break\": \"10\"}, \"busco_mode\": {\"mode\": \"geno\", \"__current_case__\": 0, \"miniprot\": true, \"use_augustus\": {\"use_augustus_selector\": \"no\", \"__current_case__\": 0}}, \"input\": {\"__class__\": \"RuntimeValue\"}, \"lineage\": {\"lineage_mode\": \"select_lineage\", \"__current_case__\": 1, \"lineage_dataset\": \"mucorales_odb10\"}, \"lineage_conditional\": {\"selector\": \"download\", \"__current_case__\": 1}, \"outputs\": [\"short_summary\", \"missing\", \"image\", \"gff\"], \"__page__\": null, \"__rerun_remap_job_id__\": null}",
- "tool_version": "5.5.0+galaxy0",
+ "tool_state": "{\"chr_replace\": {\"__class__\": \"RuntimeValue\"}, \"decode_url\": false, \"expose\": false, \"filtering\": null, \"full_gff_attribute_preservation\": false, \"gffs\": {\"gff_fmt\": \"none\", \"__current_case__\": 0}, \"input\": {\"__class__\": \"ConnectedValue\"}, \"maxintron\": null, \"merging\": {\"merge_sel\": \"none\", \"__current_case__\": 0}, \"reference_genome\": {\"source\": \"history\", \"__current_case__\": 2, \"genome_fasta\": {\"__class__\": \"ConnectedValue\"}, \"ref_filtering\": null, \"fa_outputs\": [\"-y pep.fa\"]}, \"region\": {\"region_filter\": \"none\", \"__current_case__\": 0}, \"__page__\": null, \"__rerun_remap_job_id__\": null}",
+ "tool_version": "2.2.1.4+galaxy0",
"type": "tool",
- "uuid": "af725f3e-ee5a-485b-af11-d87a8ecf9aeb",
+ "uuid": "00d60c74-1ed5-4529-aa82-8745b50205b7",
"when": null,
"workflow_outputs": [
{
- "label": "BUSCO sum",
- "output_name": "busco_sum",
- "uuid": "703a797f-428b-4c41-984f-1283bb8eecaa"
- },
- {
- "label": "BUSCO miniprot",
- "output_name": "busco_miniprot",
- "uuid": "e3879637-613a-45cf-90a0-3eecab2e0981"
- },
- {
- "label": "BUSCO gff3",
- "output_name": "busco_gff",
- "uuid": "acbfdee0-cafd-46e8-81e9-8a1fd22ee758"
- },
- {
- "label": "BUSCO summary image",
- "output_name": "summary_image",
- "uuid": "5574f029-f31d-4d20-a847-4d1818f95707"
- },
- {
- "label": "BUSCO missing",
- "output_name": "busco_missing",
- "uuid": "b19bfb5f-901f-4568-a461-53bfc980bdcc"
- },
- {
- "label": "BUSCO table",
- "output_name": "busco_table",
- "uuid": "0ba757ff-6c1b-496b-a288-335369058923"
+ "label": "gffread_pep",
+ "output_name": "output_pep",
+ "uuid": "aa178118-cd37-495b-9e81-e2e53ebf27fd"
}
]
},
"4": {
- "annotation": "Calculate statistics from a genome annotation in GFF3 format",
+ "annotation": "Generates statistics and graphs for genome annotation",
"content_id": "toolshed.g2.bx.psu.edu/repos/iuc/jcvi_gff_stats/jcvi_gff_stats/0.8.4",
"errors": null,
"id": 4,
@@ -272,16 +346,12 @@
}
},
"inputs": [
- {
- "description": "runtime parameter for tool Genome annotation statistics",
- "name": "gff"
- },
{
"description": "runtime parameter for tool Genome annotation statistics",
"name": "ref_genome"
}
],
- "label": "Genome annotation statistics Helixer",
+ "label": "Genome annotation statistics",
"name": "Genome annotation statistics",
"outputs": [
{
@@ -294,8 +364,8 @@
}
],
"position": {
- "left": 1011.9681945236466,
- "top": 0.0
+ "left": 488.25061259116643,
+ "top": 991.5198240353345
},
"post_job_actions": {},
"tool_id": "toolshed.g2.bx.psu.edu/repos/iuc/jcvi_gff_stats/jcvi_gff_stats/0.8.4",
@@ -305,26 +375,26 @@
"owner": "iuc",
"tool_shed": "toolshed.g2.bx.psu.edu"
},
- "tool_state": "{\"gff\": {\"__class__\": \"RuntimeValue\"}, \"ref_genome\": {\"genome_type_select\": \"history\", \"__current_case__\": 1, \"genome\": {\"__class__\": \"RuntimeValue\"}}, \"__page__\": null, \"__rerun_remap_job_id__\": null}",
+ "tool_state": "{\"gff\": {\"__class__\": \"ConnectedValue\"}, \"ref_genome\": {\"genome_type_select\": \"history\", \"__current_case__\": 1, \"genome\": {\"__class__\": \"ConnectedValue\"}}, \"__page__\": null, \"__rerun_remap_job_id__\": null}",
"tool_version": "0.8.4",
"type": "tool",
- "uuid": "64572fdd-797c-44e9-8662-2ae4d51528c1",
+ "uuid": "f47f89eb-23f4-4a16-b0a8-49d8e62c9f3d",
"when": null,
"workflow_outputs": [
{
- "label": "graphs helixer",
- "output_name": "graphs",
- "uuid": "a445f12b-685d-47af-b407-90fa7ea935b1"
+ "label": "summary",
+ "output_name": "summary",
+ "uuid": "fb8ed4c9-4b55-4547-880d-1916a91f8a6e"
},
{
- "label": "summary helixer",
- "output_name": "summary",
- "uuid": "8eae8a88-c73d-42dc-ad0d-62d2f1d29c6a"
+ "label": "graphs",
+ "output_name": "graphs",
+ "uuid": "4638cc23-fdb6-4e82-9cdf-c9fe38e76bd7"
}
]
},
"5": {
- "annotation": "Visualization",
+ "annotation": "JBrowse",
"content_id": "toolshed.g2.bx.psu.edu/repos/iuc/jbrowse/jbrowse/1.16.11+galaxy1",
"errors": null,
"id": 5,
@@ -353,8 +423,8 @@
}
],
"position": {
- "left": 1175.5063439719256,
- "top": 231.38681642718768
+ "left": 1065.313344724818,
+ "top": 719.9967480789329
},
"post_job_actions": {},
"tool_id": "toolshed.g2.bx.psu.edu/repos/iuc/jbrowse/jbrowse/1.16.11+galaxy1",
@@ -364,21 +434,158 @@
"owner": "iuc",
"tool_shed": "toolshed.g2.bx.psu.edu"
},
- "tool_state": "{\"action\": {\"action_select\": \"create\", \"__current_case__\": 0}, \"gencode\": \"1\", \"jbgen\": {\"defaultLocation\": \"\", \"trackPadding\": \"20\", \"shareLink\": true, \"aboutDescription\": \"\", \"show_tracklist\": true, \"show_nav\": true, \"show_overview\": true, \"show_menu\": true, \"hideGenomeOptions\": false}, \"plugins\": {\"BlastView\": true, \"ComboTrackSelector\": false, \"GCContent\": false}, \"reference_genome\": {\"genome_type_select\": \"history\", \"__current_case__\": 1, \"genome\": {\"__class__\": \"RuntimeValue\"}}, \"standalone\": \"minimal\", \"track_groups\": [{\"__index__\": 0, \"category\": \"Annotation\", \"data_tracks\": [{\"__index__\": 0, \"data_format\": {\"data_format_select\": \"gene_calls\", \"__current_case__\": 2, \"annotation\": {\"__class__\": \"RuntimeValue\"}, \"match_part\": {\"match_part_select\": false, \"__current_case__\": 1}, \"index\": false, \"track_config\": {\"track_class\": \"NeatHTMLFeatures/View/Track/NeatFeatures\", \"__current_case__\": 3, \"html_options\": {\"topLevelFeatures\": null}}, \"jbstyle\": {\"style_classname\": \"feature\", \"style_label\": \"product,name,id\", \"style_description\": \"note,description\", \"style_height\": \"10px\", \"max_height\": \"600\"}, \"jbcolor_scale\": {\"color_score\": {\"color_score_select\": \"none\", \"__current_case__\": 0, \"color\": {\"color_select\": \"automatic\", \"__current_case__\": 0}}}, \"jb_custom_config\": {\"option\": []}, \"jbmenu\": {\"track_menu\": []}, \"track_visibility\": \"default_off\", \"override_apollo_plugins\": \"False\", \"override_apollo_drag\": \"False\"}}]}], \"uglyTestingHack\": \"\", \"__page__\": null, \"__rerun_remap_job_id__\": null}",
+ "tool_state": "{\"action\": {\"action_select\": \"create\", \"__current_case__\": 0}, \"gencode\": \"1\", \"jbgen\": {\"defaultLocation\": \"\", \"trackPadding\": \"20\", \"shareLink\": true, \"aboutDescription\": \"\", \"show_tracklist\": true, \"show_nav\": true, \"show_overview\": true, \"show_menu\": true, \"hideGenomeOptions\": false}, \"plugins\": {\"BlastView\": true, \"ComboTrackSelector\": false, \"GCContent\": false}, \"reference_genome\": {\"genome_type_select\": \"history\", \"__current_case__\": 1, \"genome\": {\"__class__\": \"ConnectedValue\"}}, \"standalone\": \"minimal\", \"track_groups\": [{\"__index__\": 0, \"category\": \"Annotation\", \"data_tracks\": [{\"__index__\": 0, \"data_format\": {\"data_format_select\": \"gene_calls\", \"__current_case__\": 2, \"annotation\": {\"__class__\": \"ConnectedValue\"}, \"match_part\": {\"match_part_select\": false, \"__current_case__\": 1}, \"index\": false, \"track_config\": {\"track_class\": \"NeatHTMLFeatures/View/Track/NeatFeatures\", \"__current_case__\": 3, \"html_options\": {\"topLevelFeatures\": null}}, \"jbstyle\": {\"style_classname\": \"feature\", \"style_label\": \"product,name,id\", \"style_description\": \"note,description\", \"style_height\": \"10px\", \"max_height\": \"600\"}, \"jbcolor_scale\": {\"color_score\": {\"color_score_select\": \"none\", \"__current_case__\": 0, \"color\": {\"color_select\": \"automatic\", \"__current_case__\": 0}}}, \"jb_custom_config\": {\"option\": []}, \"jbmenu\": {\"track_menu\": []}, \"track_visibility\": \"default_off\", \"override_apollo_plugins\": \"False\", \"override_apollo_drag\": \"False\"}}]}], \"uglyTestingHack\": \"\", \"__page__\": null, \"__rerun_remap_job_id__\": null}",
"tool_version": "1.16.11+galaxy1",
"type": "tool",
- "uuid": "f77404b0-6e49-4951-9553-65ef0a24499a",
+ "uuid": "04807fae-95f6-49c1-893e-76932a79cdf9",
"when": null,
"workflow_outputs": [
{
- "label": "JBrowse output",
+ "label": "output",
"output_name": "output",
- "uuid": "2bac35a7-8379-4269-9769-e0e0c541f4a3"
+ "uuid": "19976896-9df1-45e4-9c96-89e24ae6e596"
+ }
+ ]
+ },
+ "6": {
+ "annotation": "Completeness assessment of the genome using the Busco tool",
+ "content_id": "toolshed.g2.bx.psu.edu/repos/iuc/busco/busco/5.7.1+galaxy0",
+ "errors": null,
+ "id": 6,
+ "input_connections": {
+ "input": {
+ "id": 3,
+ "output_name": "output_pep"
+ }
+ },
+ "inputs": [],
+ "label": "Busco on protein",
+ "name": "Busco",
+ "outputs": [
+ {
+ "name": "busco_sum",
+ "type": "txt"
+ },
+ {
+ "name": "busco_table",
+ "type": "tabular"
+ },
+ {
+ "name": "busco_missing",
+ "type": "tabular"
+ },
+ {
+ "name": "summary_image",
+ "type": "png"
+ },
+ {
+ "name": "busco_gff",
+ "type": "gff3"
+ }
+ ],
+ "position": {
+ "left": 1166.6977253236494,
+ "top": 58.61198039869754
+ },
+ "post_job_actions": {},
+ "tool_id": "toolshed.g2.bx.psu.edu/repos/iuc/busco/busco/5.7.1+galaxy0",
+ "tool_shed_repository": {
+ "changeset_revision": "2babe6d5c561",
+ "name": "busco",
+ "owner": "iuc",
+ "tool_shed": "toolshed.g2.bx.psu.edu"
+ },
+ "tool_state": "{\"adv\": {\"evalue\": \"0.001\", \"limit\": \"3\", \"contig_break\": \"10\"}, \"busco_mode\": {\"mode\": \"prot\", \"__current_case__\": 2}, \"input\": {\"__class__\": \"ConnectedValue\"}, \"lineage\": {\"lineage_mode\": \"auto_detect\", \"__current_case__\": 0, \"auto_lineage\": \"--auto-lineage\"}, \"lineage_conditional\": {\"selector\": \"cached\", \"__current_case__\": 0, \"cached_db\": \"v5\"}, \"outputs\": [\"short_summary\", \"image\", \"gff\", \"missing\"], \"__page__\": null, \"__rerun_remap_job_id__\": null}",
+ "tool_version": "5.7.1+galaxy0",
+ "type": "tool",
+ "uuid": "51dcc6a4-ff87-4a98-98fa-de00ce54325f",
+ "when": null,
+ "workflow_outputs": [
+ {
+ "label": "busco_gff_pep",
+ "output_name": "busco_gff",
+ "uuid": "1db166fb-10c2-4823-a80c-9f22c7c15576"
+ },
+ {
+ "label": "summary_image_pep",
+ "output_name": "summary_image",
+ "uuid": "13c6bee4-824c-4533-bc78-c99ddf0b190d"
+ },
+ {
+ "label": "busco_sum_pep",
+ "output_name": "busco_sum",
+ "uuid": "f44047d9-e713-41d9-a9f9-5543f0371d9d"
+ },
+ {
+ "label": "busco_table_pep",
+ "output_name": "busco_table",
+ "uuid": "1a113d6c-a167-432b-8200-dfb3aedc4ba1"
+ },
+ {
+ "label": "busco_missing_pep",
+ "output_name": "busco_missing",
+ "uuid": "dc2d4533-d9c2-4cb0-a144-184e90fd4e01"
+ }
+ ]
+ },
+ "7": {
+ "annotation": "OMArk",
+ "content_id": "toolshed.g2.bx.psu.edu/repos/iuc/omark/omark/0.3.0+galaxy2",
+ "errors": null,
+ "id": 7,
+ "input_connections": {
+ "input": {
+ "id": 3,
+ "output_name": "output_pep"
+ }
+ },
+ "inputs": [
+ {
+ "description": "runtime parameter for tool OMArk",
+ "name": "input_iso"
+ }
+ ],
+ "label": "OMArk",
+ "name": "OMArk",
+ "outputs": [
+ {
+ "name": "omark_detail_sum",
+ "type": "txt"
+ },
+ {
+ "name": "omark_sum",
+ "type": "sum"
+ }
+ ],
+ "position": {
+ "left": 1167.994173976809,
+ "top": 375.00649693590475
+ },
+ "post_job_actions": {},
+ "tool_id": "toolshed.g2.bx.psu.edu/repos/iuc/omark/omark/0.3.0+galaxy2",
+ "tool_shed_repository": {
+ "changeset_revision": "6f570ba54b41",
+ "name": "omark",
+ "owner": "iuc",
+ "tool_shed": "toolshed.g2.bx.psu.edu"
+ },
+ "tool_state": "{\"database\": \"Primates-v2.0.0.h5\", \"input\": {\"__class__\": \"ConnectedValue\"}, \"input_iso\": {\"__class__\": \"RuntimeValue\"}, \"omark_mode\": false, \"outputs\": \"detail_sum\", \"r\": null, \"t\": null, \"__page__\": null, \"__rerun_remap_job_id__\": null}",
+ "tool_version": "0.3.0+galaxy2",
+ "type": "tool",
+ "uuid": "75e1dde7-5d60-4092-af57-cd7b065145e2",
+ "when": null,
+ "workflow_outputs": [
+ {
+ "label": "omark_detail_sum",
+ "output_name": "omark_detail_sum",
+ "uuid": "de489b9c-8808-47d4-9384-7617c33a9d34"
}
]
}
},
- "tags": [],
- "uuid": "d2fa1512-57c4-4f45-85fe-4d4bfe801f7a",
- "version": 3
-}
+ "tags": [
+ "genome-annotation"
+ ],
+ "uuid": "7a0c9f35-37a9-404e-a307-aed30a578b0c",
+ "version": 1
+}
\ No newline at end of file
diff --git a/topics/imaging/images/omero-suite/omero_credential.png b/topics/imaging/images/omero-suite/omero_credential.png
new file mode 100644
index 0000000000000..dea14bdf717c2
Binary files /dev/null and b/topics/imaging/images/omero-suite/omero_credential.png differ
diff --git a/topics/imaging/images/omero-suite/omero_import.png b/topics/imaging/images/omero-suite/omero_import.png
new file mode 100644
index 0000000000000..4b72371b226d3
Binary files /dev/null and b/topics/imaging/images/omero-suite/omero_import.png differ
diff --git a/topics/imaging/images/omero-suite/omero_metadata.png b/topics/imaging/images/omero-suite/omero_metadata.png
new file mode 100644
index 0000000000000..731600d7dd3ce
Binary files /dev/null and b/topics/imaging/images/omero-suite/omero_metadata.png differ
diff --git a/topics/imaging/images/omero-suite/omero_rois.png b/topics/imaging/images/omero-suite/omero_rois.png
new file mode 100644
index 0000000000000..3d7b89dbfece1
Binary files /dev/null and b/topics/imaging/images/omero-suite/omero_rois.png differ
diff --git a/topics/imaging/images/omero-suite/workflow.png b/topics/imaging/images/omero-suite/workflow.png
new file mode 100644
index 0000000000000..f480d8f4ca958
Binary files /dev/null and b/topics/imaging/images/omero-suite/workflow.png differ
diff --git a/topics/imaging/images/omero-suite/workflow_invocation.png b/topics/imaging/images/omero-suite/workflow_invocation.png
new file mode 100644
index 0000000000000..af0a69f1cf50e
Binary files /dev/null and b/topics/imaging/images/omero-suite/workflow_invocation.png differ
diff --git a/topics/imaging/tutorials/detection-of-mitoflashes/data-library.yaml b/topics/imaging/tutorials/detection-of-mitoflashes/data-library.yaml
new file mode 100644
index 0000000000000..9525c4bd2572b
--- /dev/null
+++ b/topics/imaging/tutorials/detection-of-mitoflashes/data-library.yaml
@@ -0,0 +1,19 @@
+---
+destination:
+ type: library
+ name: GTN - Material
+ description: Galaxy Training Network Material
+ synopsis: Galaxy Training Network Material. See https://training.galaxyproject.org
+items:
+- name: Imaging
+ description: Image analysis using Galaxy
+ items:
+ - name: Tracking of mitochondria and capturing mitoflashes
+ items:
+ - name: 'DOI: 10.5281/zenodo.14071552'
+ description: latest
+ items:
+ - url: https://zenodo.org/record/14071552/files/mitoflashes_8bit.tiff
+ src: url
+ ext:
+ info: https://doi.org/10.5281/zenodo.14071552
diff --git a/topics/imaging/tutorials/omero-suite/data-library.yaml b/topics/imaging/tutorials/omero-suite/data-library.yaml
new file mode 100644
index 0000000000000..839bf526ef526
--- /dev/null
+++ b/topics/imaging/tutorials/omero-suite/data-library.yaml
@@ -0,0 +1,19 @@
+destination:
+ type: library
+ name: GTN - Material
+ description: Galaxy Training Network Material
+ synopsis: Galaxy Training Network Material. See https://training.galaxyproject.org
+items:
+- name: Imaging
+ description: Image management using Galaxy tools
+ items:
+ - name: Image data for OMERO
+ items:
+ - name: 'DOI: 14205500'
+ description: latest
+ items:
+ - url: https://zenodo.org/records/14205500
+ src: url
+ ext: zip
+ info: https://zenodo.org/records/14205500
+
diff --git a/topics/imaging/tutorials/omero-suite/tutorial.md b/topics/imaging/tutorials/omero-suite/tutorial.md
new file mode 100644
index 0000000000000..a99793075468e
--- /dev/null
+++ b/topics/imaging/tutorials/omero-suite/tutorial.md
@@ -0,0 +1,376 @@
+---
+layout: tutorial_hands_on
+title: Overview of the Galaxy OMERO-suite - Upload images and metadata in OMERO using Galaxy
+level: Intermediate
+zenodo_link: https://zenodo.org/records/14205500
+questions:
+- How can I use Galaxy to upload images and metadata into OMERO?
+- Which are the different tools of the Galaxy OMERO-suite?
+objectives:
+- Understand how the different tools of the Galaxy OMERO-suite works
+- Build a complete pipeline for image and metadata upload into OMERO
+time_estimation: 1H
+key_points:
+- Proper data management plays an important role in image processing
+- Galaxy can facilitate the integration of data management tools into an image processing pipeline
+- The OMERO-suite allows to easily manage by building a FAIR pipeline
+contributions:
+ authorship:
+ - rmassei
+ editing:
+ - bgruening
+ funding:
+ - nfdi4bioimage
+---
+The efficient and accurate treatment of microscopy metadata is of great importance, as it
+provides insights that are essential for effective image management, search, organisation,
+interpretation, and sharing. Considering this, it is vital to find ways to properly deal with the huge amount
+of complex and unstructured data for implementing **[Findable, Accessible,
+Interoperable and Reusable (FAIR)](https://www.nature.com/articles/sdata201618)** concepts in bio-imaging.
+
+One of the most flexible and used open-source tools for image and metadata management can be
+identified as **[OMERO
+(Open Microscopy Environment Remote Objects)](https://www.nature.com/articles/nmeth.1896)**. **OMERO** is an open-source
+software platform designed to manage, visualise, and analyse large sets of biological image
+data. Developed by the Open Microscopy Environment consortium, **OMERO** provides researchers
+with a centralised repository to store images and metadata, tools for collaborative
+sharing, and advanced functionalities for image processing and analysis.
+
+In this tutorial, you will learn how to use the different tools of the **Galaxy OMERO-suite**.
+The **Galaxy OMERO-suite** is based on the Python packages omero-py and ezomero, and it allows interactively building pipelines to upload and fetch image data in OMERO
+using a Galaxy workflow.
+Images can automatically be enriched with metadata (i.e. key-value pairs, tags, raw data, regions
+of interest) and uploaded to an OMERO server. The tools give the possibility
+to the user to intuitively fetch images from the local server and
+perform image analysis.
+
+>
+>
+> In this tutorial, we will cover:
+>
+> 1. Learn the different tools of the Galaxy OMERO-suite to manage image data and metadata
+> 2. Get data from an OMERO instance using the Galaxy OMERO-suite
+> 3. Build a pipeline for image and metadata upload into OMERO
+{: .agenda}
+
+
+## Before Starting - Set up the OMERO credentials
+Before starting you need to set up your OMERO credential.
+This is necessary to connect to a target OMERO instance and needs to be performed just one time.
+
+The OMERO credentials have to be saved in your preferences
+(*User → Preferences → Manage information*).
+
+
+
+This feature is embedded in Galaxy and needs to be enabled by the admin
+of the server you are using. You can now use it in [UseGalaxy.eu](https://usegalaxy.eu/) or ask your
+admin to add it. For more info about the configuration on the admin side,
+please take a look at the [README file of the tool](https://github.com/galaxyproject/tools-iuc/blob/main/tools/idr_download/README.md).
+
+**Please Notice**: The configuration file, which contains your OMERO password and username, will be stored in the job working directory.
+ This directory only exists during the runtime of the job and should only be accessible by the system user that runs the job.
+ However, please be aware that your username and password **may be exposed** to users with administrative rights.
+ We are working on increasing the security of the OMERO suite
+
+# Prepare your data
+
+We need a dataset to upload into your OMERO instance.
+To this end, we have prepared an integrative dataset with images, metadata and regions of interest (ROIs) in Zenodo.
+
+
+## Data Upload - Image, metadata and ROI files
+
+> Data Upload
+>
+> 1. Create a new history for this tutorial in Galaxy.
+>
+> {% snippet faqs/galaxy/histories_create_new.md %}
+>
+> 2. Import the nuclei imaging data from [Zenodo](https://zenodo.org/records/14196675) or
+> from the shared data library:
+> - **Important:** Choose the correct data type if prompted.
+>
+> ```
+> https://zenodo.org/records/14205500
+> ```
+>
+> {% snippet faqs/galaxy/datasets_import_via_link.md %}
+>
+> {% snippet faqs/galaxy/datasets_import_from_data_library.md %}
+>
+>
+> - Select the following files:
+>
+> - `image85-H-01-00.tif`
+> - `image85-H-01-00.tif.tabular`
+> - `image86-H-02-00.tif`
+> - `image86-H-02-00.tif.tabular`
+> - `image87-H-03-00.tif`
+> - `image87-H-03-00.tif.tabular`
+> - `metadata_dataset.csv`
+>
+> 3. Tag each dataset with a label like "image" (tif files), "rois" (tabular files) and "metadata" (CSV file)
+> for easy identification
+{: .hands_on}
+
+# Upload images and metadata
+
+In this section, we will focus on how uploading images and metadata into a user-defined OMERO instance.
+This is done by using the **OMERO Image Import** and **OMERO Metadata Import** tools.
+
+## Step 1: Image Upload
+
+> Upload images into OMERO
+>
+> 1. {% tool [OMERO Image Import](toolshed.g2.bx.psu.edu/repos/ufz/omero_import/omero_import/5.18.0+galaxy3) %} with the following recommended parameters:
+> - {% icon param-file %} *Images to import into OMERO*: You can select here different files to import into OMERO.
+> Select the datasets `image85-H-01-00.tif`, `image86-H-02-00.tif` and `image87-H-03-00.tif`
+> - **OMERO host URL**: Input the URL of your OMERO instance.
+> - **OMERO port**: Input the OMERO port (pre-defined value, *4064*)
+> - {% icon param-file %} **Target Dataset Name**: Type "My_OMERO_Test"
+>
+> A log file text file will be
+> created with the OMERO ID of the newly imported images
+{: .hands_on}
+
+After tool execution, you can check the images in your OMERO instance!
+
+
+Obviously, the Dataset and Image ID change according to your instance... In this case, we have created a new
+dataset called "My_OMERO_Test" with ID 4005, while images have IDs 30781, 30782 and 30783.
+
+## Step 2: Upload metadata
+
+You can upload two different kinds of metadata files using the OMERO-suite.
+
+- **Key Value Pairs:** Useful for displaying dataset and image metadata in OMERO.web.
+Key Value pairs are for unstructured data where every object can have different keys, and they can be easily edited ad-hoc by users.
+- **Tables:** Useful for showing results for multiple images at the same time. Tables are suitable for e.g. analysis results or large structured data, columns have defined types (kv pairs are all strings) and can be queried like a database table.
+
+We will learn now how to upload **Key-Value pairs** using the **OMERO Metadata Import** tool
+
+> Upload Key-Value Pairs into OMERO
+>
+> 1. {% tool [OMERO Metadata Import](toolshed.g2.bx.psu.edu/repos/ufz/omero_metadata_import/omero_metadata_import/5.18.0+galaxy3) %} with the following recommended parameters:
+> - {% icon param-file %} *Annotation file*: `metadata_file.tsv`
+> - **OMERO host URL**: Input the URL of your OMERO instance.
+> - **OMERO port**: Input the OMERO port (pre-defined value, *4064*)
+> - {% icon param-file %} **Target Object Type**: Select *"Dataset"*
+> - **Selection**: Select *"Target an existing object"*
+> - **Object ID**: Input the ID of the previously created dataset. *4005* for this training
+> - **Annotation type**: Select *"KV"*
+> - **Annotation Name**: Type *"REMBI_Annotation"*
+{: .hands_on}
+
+Switch back to your OMERO instance.
+The Key Values are now in OMERO, associated with a target dataset! Well done!
+
+
+# Upload region of interest (ROIs)
+OMERO support the ROI visualization in OMERO.viewer.
+In this section you will learn to associate ROIs coordinates to
+an image stored in OMERO using the **OMERO ROI Import** tool.
+
+Please note that this tool supports just polygon ROIs given in a specific format.
+The tool [Analyze particles](https://toolshed.g2.bx.psu.edu/view/imgteam/imagej2_analyze_particles_binary/862af85a50ec)
+can automatically create the ROIs in this specific format.
+
+Check the `roi_file.tsv` to have an example. Everything is explained in the tools help section!
+
+
+> Upload images into OMERO
+>
+> 1. {% tool [OMERO ROI Import](toolshed.g2.bx.psu.edu/repos/ufz/omero_roi_import/omero_roi_import/5.18.0+galaxy4) %} with the following recommended parameters:
+> - {% icon param-file %} **Tab File with ROIs**: `roi_file.tsv`
+> - **OMERO host URL**: Input the URL of your OMERO instance.
+> - **OMERO port**: Input the OMERO port (pre-defined value, *4064*)
+> - **Image ID where annotate the ROIs**: Select the **image ID** where to annotate the ROIs. We will go for *30782*
+{: .hands_on}
+
+Switch back to your OMERO instance and the image by double-clicking it.
+This will open the OMERO.viewer.
+Go now to the "ROIs" sub-panel.
+The ROIs are now annotated in OMERO and associated with the target image! **Awesome!**
+
+
+
+
+# Get OMERO object IDs and Annotations
+You can get information on projects, datasets and image IDs.
+Furthermore, you can fetch annotations and tables
+associated with an OMERO object. This is done with the **OMERO get IDs** and
+**OMERO get Object** tools, respectively.
+
+First of all, let's try to get all the image IDs present in all Datasets!
+
+> Upload images into OMERO
+>
+> 1. {% tool [OMERO get IDs](toolshed.g2.bx.psu.edu/repos/ufz/omero_get_id/omero_get_id/5.18.0+galaxy0) %} with the following recommended parameters:
+> - **OMERO host URL**: Input the URL of your OMERO instance.
+> - **OMERO port**: Input the OMERO port (pre-defined value, *4064*)
+> - **Type of object to fetch ID:**: Select *"Dataset IDs"*
+> - **Which datasets?**: Select *"All datasets"*
+> - **ID of the project**: *0*
+{: .hands_on}
+
+Since we have just one dataset in your OMERO instance, the tool will produce a tabular file
+with the IDs of the three images we just uploaded.
+
+| 1 |
+|-------|
+| 30781 |
+| 30782 |
+| 30783 |
+
+Now, let's try to get an annotation file...
+
+> Get an annotation file from OMERO
+>
+> 1. {% tool [OMERO get Object](toolshed.g2.bx.psu.edu/repos/ufz/omero_get_value/omero_get_value/5.18.0+galaxy0) %} with the following recommended parameters:
+> - **OMERO host URL**: The target OMERO host URL
+> - **OMERO port**: The OMERO port, pre-defined to *4064*
+> - **Type of object to fetch:**: Select *"Annotation"*
+> - **How do you provide the ID(s) of the OMERO object?**: Select *"Comma separated values"*
+> - **ID(s) of the object(s) to fetch on OMERO separated by comma**: 4005
+{: .hands_on}
+
+Perfect, you fetched the Annotation file "REMBI_Annotation".
+**OMERO get Object** is particularly useful when you want to fetch data associated with your dataset or
+images!
+
+# Filter OMERO object based on filename, tags and Key-Value Pairs
+Finally, you filter OMERO objects based on their features... One option is to filter by filename,
+a useful parameter to apply if you want to get certain images.
+
+This can be done by using the **OMERO IDs** Tool
+
+> Upload images into OMERO
+>
+> 1. {% tool [OMERO IDs](toolshed.g2.bx.psu.edu/repos/ufz/omero_filter/omero_filter/5.18.0+galaxy0) %} with the following recommended parameters:
+> - **OMERO host URL**: The target OMERO host URL
+> - **OMERO port**: The OMERO port, pre-defined to *4064*
+> - **Filename to search among the image IDs**: Type `image85-H-01-00_tif.tiff`
+> - **List of images IDs**: Type *30781,30782,30783*
+{: .hands_on}
+
+As an output, you will have a tabular file with the following info:
+
+| 1 |
+|-------|
+| 30782 |
+
+Which is ID associated to the image with file name "image85-H-01-00_tif.tiff"!
+
+However, you can also use this tool to fetch images with specific file names or tags,
+making data fetching an easy task...
+
+# A full workflow for data management with the OMERO suite
+
+In this section, you will learn to integrate all the different tools to build a workflow for uploading
+an images, metadata and ROIs into OMERO.
+
+## Step 1 - Define the OMERO inputs
+To make the workflow work, you need to define six inputs.
+
+This will make this workflow reusable with different datasets.
+
+> Create the inputs for the OMERO pipeline
+>
+> 1. Create a new workflow in the workflow editor.
+>
+> {% snippet faqs/galaxy/workflows_create_new.md %}
+>
+> 2. Select {% icon tool %} **Input dataset collection** from the list of tools:
+> - {% icon param-collection %} **1: Input dataset collection** appears in your workflow.
+> Change the "Label" of this input to *Input image Dataset*.
+> 3. Add two {% icon tool %} **Input dataset**:
+> - {% icon param-file %} **2: Input Dataset** and {% icon param-file %} **3: Input Dataset** appears in your workflow.
+> Change the "Label" of these inputs to *Input Tabular ROIs* and *Input Metadata File*
+> 4. Add three {% icon tool %}**Simple Input for Workflow Logic**:
+> - **4: Simple input for workflow logic**, **5: Simple input for workflow logic**,
+> **6: Simple input for workflow logic** appear in your workflow.
+> Change the "Label" of these inputs to *Target Dataset Name*, *OMERO instance address* and
+> *Annotation type*
+{: .hands_on}
+
+## Step 2 - OMERO Import
+We now add the step for the image import
+
+> Add the image upload step to the workflow
+>
+>1. While in the workflow editor add {% tool [OMERO Image Import](toolshed.g2.bx.psu.edu/repos/ufz/omero_import/omero_import/5.18.0+galaxy3) %} from the list of tools:
+> - Connect the output of {% icon param-file %} **1: Input image Datasets** to the "Images to Import in OMERO"
+> input of {% icon tool %} **7: OMERO Image Import**.
+> - Connect the output of {% icon param-file %} **4: Target Dataset Name** to the "Target Dataset Name"
+> input of {% icon tool %} **7: OMERO Image Import**.
+> - Connect the output of {% icon param-file %} **5: OMERO instance address** to the "OMERO host url"
+> input of {% icon tool %} **7: OMERO Image Import**.
+{: .hands_on}
+
+## Step 3 - Dynamically parse image IDs
+We need now a small series of steps to get the ID of the image we just upload into OMERO
+This can be done with the following tool sequence:
+
+> Parse Images IDs
+>
+>1. Add {% icon tool %} **Convert** from the list of tools:
+> - Connect the output of {% icon tool %} **7: OMERO Image Import** to the {% icon tool %} **8: Convert** input
+>2. Add {% icon tool %} **Replace text** from the list of tools:
+> - Connect the output of {% icon tool %} **8: Convert** to the {% icon tool %} **Replace text** input
+>3. Add {% icon tool %} **Split File** from the list of tools:
+> - Connect the output of {% icon tool %} **9: Replace text** to the {% icon tool %} **10: Split File**
+>4. Add {% icon tool %} **Parse parameter value** from the list of tools:
+> - Connect the output of {% icon tool %} **10: Split File** to the {% icon tool %} **11: Parse parameter value**
+{: .hands_on}
+
+We got the OMERO image ID! We can now use it as an input for next sequence of tools...
+
+## Step 4 - Metadata and ROIs input
+
+The last section is to upload metadata and ROIs
+
+> Add tools to upload metadata and ROIs
+>
+>1. Add {% tool [OMERO Metadata Import](toolshed.g2.bx.psu.edu/repos/ufz/omero_metadata_import/omero_metadata_import/5.18.0+galaxy3) %} from the list of tools:
+> - Connect the output of {% icon param-file %} **5: OMERO instance address** to the "OMERO host url"
+> input of {% icon tool %} **12: OMERO Metadata Import**.
+> - Connect the output of {% icon param-file %} **11: Parse parameter value** to the "Object ID"
+> input of {% icon tool %} **12: OMERO Metadata Import**.
+> - Connect the output of {% icon param-file %} **6: Annotation type** to the "Annotation type"
+> input of {% icon tool %} **12: OMERO Metadata Import**.
+> - Connect the output of {% icon param-file %} **3: Input Metadata File** to the "Annotation file"
+> input of {% icon tool %} **12: OMERO Metadata Import**.
+>2. Add {% tool [OMERO ROI Import](toolshed.g2.bx.psu.edu/repos/ufz/omero_roi_import/omero_roi_import/5.18.0+galaxy4) %} from the list of tools:
+> - Connect the output of {% icon param-file %} **5: OMERO instance address** to the "OMERO host url"
+> input of {% icon tool %} **13: OMERO ROI Import**.
+> - Connect the output of {% icon param-file %} **2: Input Tabular ROIs** to the "Tab File with ROIs"
+> input of {% icon tool %} **13: OMERO ROI Import**.
+> - Connect the output of {% icon param-file %} **11: Parse parameter value** to the "Image ID where annotate the ROIs"
+> input of {% icon tool %} **13: OMERO ROI Import**.
+>3. Save your workflow and name it `OMERO_basic_upload`.
+{: .hands_on}
+
+
+You are done! The final workflow should look like this:
+
+
+And you can easily add all parameters during the workflow invocation:
+
+
+This workflow can be easily re-used or turned into a sub-workflow for an image processing
+pipeline.
+
+# Conclusion
+
+In this exercise, you imported images into OMERO using Galaxy. You also learn how to
+import metadata and ROIs, as well as get information such as image ID and annotation file
+from a target OMERO instance.
+
+
+# References
+
+- Allan, C., Burel, JM., Moore, J. et al. OMERO: flexible, model-driven data management for experimental biology.
+Nat Methods 9, 245–253 (2012). https://doi.org/10.1038/nmeth.1896
+- Wilkinson, M., Dumontier, M., Aalbersberg, I. et al. The FAIR Guiding Principles for Scientific Data Management and
+stewardship. Sci Data 3, 160018 (2016). https://doi.org/10.1038/sdata.2016.18
diff --git a/topics/imaging/tutorials/omero-suite/workflows/index.md b/topics/imaging/tutorials/omero-suite/workflows/index.md
new file mode 100644
index 0000000000000..e092e0ae66ddd
--- /dev/null
+++ b/topics/imaging/tutorials/omero-suite/workflows/index.md
@@ -0,0 +1,3 @@
+---
+layout: workflow-list
+---
diff --git a/topics/introduction/images/101_11.png b/topics/introduction/images/101_11.png
index 25363ea1b40f9..e36615d3a76f9 100644
Binary files a/topics/introduction/images/101_11.png and b/topics/introduction/images/101_11.png differ
diff --git a/topics/introduction/tutorials/galaxy-intro-101-everyone/tutorial.md b/topics/introduction/tutorials/galaxy-intro-101-everyone/tutorial.md
index dc8e8c7d90177..9d8ee08c16a8b 100644
--- a/topics/introduction/tutorials/galaxy-intro-101-everyone/tutorial.md
+++ b/topics/introduction/tutorials/galaxy-intro-101-everyone/tutorial.md
@@ -29,7 +29,7 @@ contributions:
- annefou
- nagoue
- chrisbarnettster
- - michelemaroni89
+ - michelemaroni
- olanag1
- tnabtaf
- shiltemann
diff --git a/topics/introduction/tutorials/introduction/slides.html b/topics/introduction/tutorials/introduction/slides.html
index a0ee299559095..a8d2bbeabff14 100644
--- a/topics/introduction/tutorials/introduction/slides.html
+++ b/topics/introduction/tutorials/introduction/slides.html
@@ -14,7 +14,7 @@
- nsoranzo
- hexylena
- chrisbarnettster
- - michelemaroni89
+ - michelemaroni
- annefou
- nagoue
- olanag1
diff --git a/topics/microbiome/tutorials/beer-data-analysis/tutorial.md b/topics/microbiome/tutorials/beer-data-analysis/tutorial.md
index 651349a30989a..3f5b45aba42d3 100644
--- a/topics/microbiome/tutorials/beer-data-analysis/tutorial.md
+++ b/topics/microbiome/tutorials/beer-data-analysis/tutorial.md
@@ -578,7 +578,6 @@ The species identified for Chimay beers are (from the most abundant to the least
- *Saccharomyces cerevisiae*
- *Saccharomyces mikatea*: a species generally used in winemaking ({% cite bellon2013introducing %})
- *Kazachstania martiniae*: *Kazachstania* is a genus from the family Saccharomycetaceaethe.
-
- *Saccharomyces kudriavzevii*
- *Brettanomyces bruxellensis*
diff --git a/topics/microbiome/tutorials/dada-16S/tutorial.md b/topics/microbiome/tutorials/dada-16S/tutorial.md
index c19e76fe51e55..02f69e337fe42 100644
--- a/topics/microbiome/tutorials/dada-16S/tutorial.md
+++ b/topics/microbiome/tutorials/dada-16S/tutorial.md
@@ -130,7 +130,7 @@ To speed up analysis for this tutorial, we will use only a subset of this data.
Any analysis should get its own Galaxy history. So let's start by creating a new one:
-> Data upload
+> History creation
>
> 1. Create a new history for this analysis
>
diff --git a/topics/proteomics/tutorials/clinical-mp-1-database-generation/tutorial.md b/topics/proteomics/tutorials/clinical-mp-1-database-generation/tutorial.md
index 2fe9e717113a8..15c12862c8129 100644
--- a/topics/proteomics/tutorials/clinical-mp-1-database-generation/tutorial.md
+++ b/topics/proteomics/tutorials/clinical-mp-1-database-generation/tutorial.md
@@ -48,7 +48,7 @@ recordings:
- katherine-d21
---
-Metaproteomics is the large-scale characterization of the entire complement of proteins expressed by microbiota. However, metaproteomics analysis of clinical samples is challenged by the presence of abundant human (host) proteins which hampers the confident detection of lower abundant microbial proteins {% cite Batut2018 %} ; [{% cite Jagtap2015 %} .
+Metaproteomics is the large-scale characterization of the entire complement of proteins expressed by microbiota. However, metaproteomics analysis of clinical samples is challenged by the presence of abundant human (host) proteins which hampers the confident detection of lower abundant microbial proteins {% cite Batut2018 %} ; {% cite Jagtap2015 %} .
To address this, we used tandem mass spectrometry (MS/MS) and bioinformatics tools on the Galaxy platform to develop a metaproteomics workflow to characterize the metaproteomes of clinical samples. This clinical metaproteomics workflow holds potential for general clinical applications such as potential secondary infections during COVID-19 infection, microbiome changes during cystic fibrosis as well as broad research questions regarding host-microbe interactions.
@@ -177,7 +177,7 @@ For this tutorial, a literature survey was conducted to obtain 118 taxonomic spe
## Merging databases to obtain a large comprehensive database for MetaNovo
Once generated, the Species UniProt database (~3.38 million sequences) will be merged with the Human SwissProt database (reviewed only; ~20.4K sequences) and contaminant (cRAP) sequences database (116 sequences) and filtered to generate the large comprehensive database (~2.59 million sequences). The large comprehensive database will be used to generate a compact database using MetaNovo, which is much more manageable.
-> Download contaminants with **Protein Database Downloader
+> Download contaminants with **Protein Database Downloader**
>
> 1. {% tool [Protein Database Downloader](toolshed.g2.bx.psu.edu/repos/galaxyp/dbbuilder/dbbuilder/0.3.4) %} with the following parameters:
> - *"Download from?"*: `cRAP (contaminants)`
diff --git a/topics/proteomics/tutorials/clinical-mp-2-discovery/tutorial.md b/topics/proteomics/tutorials/clinical-mp-2-discovery/tutorial.md
index 2c9ade8468656..b48723580ba32 100644
--- a/topics/proteomics/tutorials/clinical-mp-2-discovery/tutorial.md
+++ b/topics/proteomics/tutorials/clinical-mp-2-discovery/tutorial.md
@@ -487,7 +487,7 @@ MaxQuant is an MS-based proteomics platform that is capable of processing raw da
>
>
> 1. What is the Experimental Design file for MaxQuant?
-> >
+>
> >
> >
> > 1. In MaxQuant, the **Experimental Design** file is used to specify the experimental conditions, sample groups, and the relationships between different samples in a proteomics experiment. This file is a crucial component of the MaxQuant analysis process because it helps the software correctly organize and analyze the mass spectrometry data. The **Experimental Design** file typically has a ".txt" extension and is a tab-delimited text file. Here's what you might include in an Experimental Design file for MaxQuant: **Sample Names** (You specify the names of each sample in your experiment. These names should be consistent with the naming conventions used in your raw data files.), **Experimental Conditions** (You define the experimental conditions or treatment groups associated with each sample. For example, you might have control and treated groups, and you would assign the appropriate condition to each sample.), **Replicates** (You indicate the replicates for each sample, which is important for assessing the statistical significance of your results. Replicates are typically denoted by numeric values (e.g., "1," "2," "3") or by unique identifiers (e.g., "Replicate A," "Replicate B")), **Labels** (If you're using isobaric labeling methods like TMT (Tandem Mass Tag) or iTRAQ (Isobaric Tags for Relative and Absolute Quantitation), you specify the labels associated with each sample. This is important for quantification.), **Other Metadata** (You can include additional metadata relevant to your experiment, such as the biological source, time points, or any other information that helps describe the samples and experimental conditions.)
diff --git a/topics/proteomics/tutorials/clinical-mp-3-verification/tutorial.md b/topics/proteomics/tutorials/clinical-mp-3-verification/tutorial.md
index a75ecd0060cc0..d2c61e7ca7fd8 100644
--- a/topics/proteomics/tutorials/clinical-mp-3-verification/tutorial.md
+++ b/topics/proteomics/tutorials/clinical-mp-3-verification/tutorial.md
@@ -105,8 +105,8 @@ Interestingly, the PepQuery tool does not rely on searching peptides against a r
>
{: .hands_on}
-# Import Workflow
+# Import Workflow
> Running the Workflow
>
@@ -304,7 +304,8 @@ We will use the Query Tabular tool {% cite Johnson2019 %} to search the PepQuery
> > SQL Query information
> > The query input files are the list of peptides and the peptide report we obtained from MaxQuant and SGPS. The query is matching each peptide (m.pep) from the PepQuery results to the peptide reports so that each verified peptide has its protein/protein group assigned to it.
> {: .comment}
->
+{: .hands_on}
+
> Remove Header with Remove beginning
>
> 1. {% tool [Remove beginning](Remove beginning1) %} with the following parameters:
@@ -363,8 +364,8 @@ Again, we will use the Query Tabular tool to retrieve UniProt IDs (accession num
> - *"Use first line as column names"*: `Yes`
> - *"Specify Column Names (comma-separated list)"*: `pep,prot`
> ` *"SQL Query to generate tabular output"*: `SELECT distinct(prot) AS Accession
-> from t1`
-> *"include query result column headers"*: `No`
+> from t1`
+> - *"include query result column headers"*: `No`
>
>
{: .hands_on}
diff --git a/topics/proteomics/tutorials/clinical-mp-4-quantitation/tutorial.md b/topics/proteomics/tutorials/clinical-mp-4-quantitation/tutorial.md
index 2f9cdaab2cf8d..f21514bbb46b4 100644
--- a/topics/proteomics/tutorials/clinical-mp-4-quantitation/tutorial.md
+++ b/topics/proteomics/tutorials/clinical-mp-4-quantitation/tutorial.md
@@ -96,11 +96,10 @@ In this current workflow, we perform Quantification using the MaxQuant tool and
> 6. Create a dataset of the RAW files.
>
> {% snippet faqs/galaxy/datasets_add_tag.md %}
->
{: .hands_on}
-# Import Workflow
+# Import Workflow
> Running the Workflow
>
@@ -123,7 +122,7 @@ In this current workflow, we perform Quantification using the MaxQuant tool and
In the [Discovery Module](https://github.com/subinamehta/training-material/blob/main/topics/proteomics/tutorials/clinical-mp-discovery/tutorial.md), we used MaxQuant to identify peptides for verification. Now, we will again use MaxQuant to further quantify the PepQuery-verified peptides, both microbial and human. More information about quantitation using MaxQuant is available, including [Label-free data analysis](https://gxy.io/GTN:T00218) and [MaxQuant and MSstats for the analysis of TMT data](https://gxy.io/GTN:T00220).
-The outputs we are most interested in consist of the `MaxQuant Evidence file`, `MaxQuant Protein Group`s, and `MaxQuant Peptides`. The `MaxQuant Peptides` file will allow us to group them to generate a list of quantified microbial peptides.
+The outputs we are most interested in consist of the `MaxQuant Evidence file`, `MaxQuant Protein Groups`, and `MaxQuant Peptides`. The `MaxQuant Peptides` file will allow us to group them to generate a list of quantified microbial peptides.
> Quantify verified peptides (from PepQuery2)
>
diff --git a/topics/sequence-analysis/metadata.yaml b/topics/sequence-analysis/metadata.yaml
index ff22e1ba3908e..bde7c320097d0 100644
--- a/topics/sequence-analysis/metadata.yaml
+++ b/topics/sequence-analysis/metadata.yaml
@@ -16,6 +16,11 @@ editorial_board:
- bebatut
- joachimwolff
+subtopics:
+ - id: basics
+ title: "The Basics"
+ description: "These tutorials cover basic operations common to many sequence-bases analyses"
+
references:
-
authors: "SciLifeLab"
diff --git a/topics/sequence-analysis/tutorials/mapping/tutorial.md b/topics/sequence-analysis/tutorials/mapping/tutorial.md
index 8ad00be2752e9..b15e2a645a2df 100644
--- a/topics/sequence-analysis/tutorials/mapping/tutorial.md
+++ b/topics/sequence-analysis/tutorials/mapping/tutorial.md
@@ -15,6 +15,7 @@ key_points:
- Know your data!
- Mapping is not trivial
- There are many mapping algorithms, it depends on your data which one to choose
+subtopic: basics
requirements:
- type: internal
topic_name: sequence-analysis
@@ -49,9 +50,9 @@ recordings:
length: 24M
galaxy_version: 24.1.2.dev0
date: '2024-09-07'
- speakers:
+ speakers:
- DinithiRajapaksha
- captioners:
+ captioners:
- DinithiRajapaksha
bot-timestamp: 1725707919
@@ -142,7 +143,7 @@ Currently, there are over 60 different mappers, and their number is growing. In
>
> 2. Inspect the `mapping stats` file by clicking on the {% icon galaxy-eye %} (eye) icon
>
->
+>
>
{: .hands_on}
diff --git a/topics/sequence-analysis/tutorials/quality-control/slides.html b/topics/sequence-analysis/tutorials/quality-control/slides.html
index 84daf4aa3b0a3..be55e34ca8547 100644
--- a/topics/sequence-analysis/tutorials/quality-control/slides.html
+++ b/topics/sequence-analysis/tutorials/quality-control/slides.html
@@ -28,6 +28,8 @@
- lleroi
- r1corre
- stephanierobin
+ editing:
+ - Swathi266
funding:
- gallantries
@@ -182,7 +184,17 @@
### Quality score encoding
+.image-100[

+]
+
+
+.pull-bottom[
+- Quality score (Q) is encoded as [ASCII characters](https://www.ascii-code.com/characters)
+- Formula to find the probability of error \\(P = 10 ^{-Q/10}\\)
+- [ASCII base 33](https://drive5.com/usearch/manual/quality_score.html) is now almost universally used. ASCII 33 is the first “normal" ASCII character.
+
+]
???
diff --git a/topics/sequence-analysis/tutorials/quality-control/tutorial.md b/topics/sequence-analysis/tutorials/quality-control/tutorial.md
index bacc51eef9f38..958dd5f142350 100644
--- a/topics/sequence-analysis/tutorials/quality-control/tutorial.md
+++ b/topics/sequence-analysis/tutorials/quality-control/tutorial.md
@@ -19,6 +19,7 @@ follow_up_training:
- mapping
time_estimation: 1H30M
level: Introductory
+subtopic: basics
key_points:
- Perform quality control on every dataset before running any other bioinformatics
analysis
@@ -36,6 +37,8 @@ contributions:
- r1corre
- stephanierobin
- neoformit
+ editing:
+ - Swathi266
funding:
- gallantries
@@ -148,12 +151,19 @@ It means that the fragment named `@M00970` corresponds to the DNA sequence `GTGC
>
> 1. Which ASCII character corresponds to the worst Phred score for Illumina 1.8+?
> 2. What is the Phred quality score of the 3rd nucleotide of the 1st sequence?
-> 3. What is the accuracy of this 3rd nucleotide?
+> 3. How to calculate the accuracy of the nucleotide base with the ASCII code `/`?
+> 4. What is the accuracy of this 3rd nucleotide?
>
> >
> > 1. The worst Phred score is the smallest one, so 0. For Illumina 1.8+, it corresponds to the `!` character.
> > 2. The 3rd nucleotide of the 1st sequence has a ASCII character `G`, which correspond to a score of 38.
-> > 3. The corresponding nucleotide `G` has an accuracy of almost 99.99%
+> > 3. This can be calculated as follows:
+> > - ASCII code for `/` is 47
+> > - Quality score = 47-33=14
+> > - Formula to find the probability of error: \\(P = 10^{-Q/10}\\)
+> > - Probability of error = \\(10^{-14/10}\\) = 0.03981
+> > - Therefore Accuracy = 100 - 0.03981 = 99.96%
+> > 4. The corresponding nucleotide `G` has an accuracy of almost 99.96%
> >
> {: .solution }
{: .question}
diff --git a/topics/single-cell/index.md b/topics/single-cell/index.md
index a7d9d0ba06f5f..3974bf1884dc5 100644
--- a/topics/single-cell/index.md
+++ b/topics/single-cell/index.md
@@ -7,9 +7,18 @@ topic_name: single-cell
## Want to explore analysis beyond our tutorials?
-Check out workflows shared by users like you!
-
-
+