In molecular dynamics (MD) simulations, the deep computational graph spanning the entire trajectory incurs significant temporal and computational costs. This limitation can be circumvented through trajectory reweighting schemes. In DMFF, the reweighting algorithm is incorporated into the MBAR method, extending the differentiable estimators for average properties and free energy. Although differentiable estimation of dynamic properties remains a challenge, introducing the reweighted MBAR estimator has largely eased the fitting of thermodynamic properties.
In the MBAR theory, it is assumed that there are K ensembles defined by (effective) potential energies
For each ensemble, the Boltzmann weight, partition function, and probability function are defined as:
For each ensemble
Within the context of MBAR, for any ensemble K, the weighted average of the observable is defined as:
To compute the average of a physical quantity
Thus, the ensemble average of A is:
Thus, the MBAR theory provides a method to estimate the ensemble averages using multiple samples from different ensembles.
In the MBAR framework,
In the reweighted MBAR estimator, we define two types of ensembles: the sampling ensemble, from which all samples are extracted (labeled as
When resample happens, Eqn. (4) is solved iteratively using standard MBAR to update
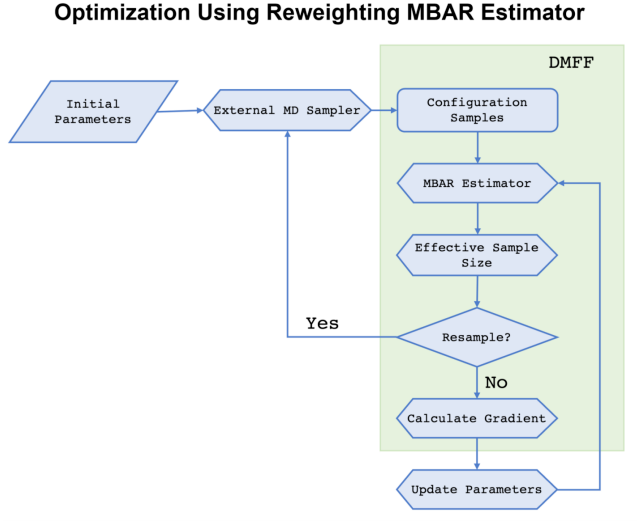
Below, we illustrate the workflow of how to use MBAR Estimator in DMFF through a case study.
If all sampling ensembles are defined as a single ensemble
and define:
then:
Refers to equations above, this equation indicates that the trajectory reweighting algorithm is a special case of the reweighted MBAR estimator.
In DMFF, when calculating the average of the physical quantity A, the formula is expressed as:
where
When
Here is a graphical representation of the workflow mentioned above:
[1]Wang X, Li J, Yang L, et al.(2022) DMFF: An Open-Source Automatic Differentiable Platform for Molecular Force Field Development and Molecular Dynamics Simulation[J].
[2]Thaler, S.; Zavadlav, J.(2021) Learning neural network potentials from experimental datavia Differentiable Trajectory Reweighting.Nature Communications, 12, 6884,
[3]Shirts, M. R.; Chodera, J. D.(2008) Statistically optimal analysis of samples from multiple equilibrium states.The Journal of Chemical Physics, 129, 124105
Function buildTrajEnergyFunction:
- Constructs a function that calculates energy for each frame in a trajectory based on a given potential function and other parameters.
- Uses neighbor lists for periodic boundary conditions.
Class TargetState:
- Represents a state of a system with a specified temperature.
- Contains methods for calculating energy of trajectories.
Class SampleState:
- A generic state from which samples (e.g., trajectories) can be taken.
- Methods allow energy calculations for each frame in a trajectory.
Class OpenMMSampleState:
- Inherits from
SampleStateand specializes in samples simulated using OpenMM.
Class Sample:
- Represents a sampled trajectory and the state from which it was sampled.
- Can generate energies of the trajectory with respect to different states.
Class ReweightEstimator:
- Estimates the weights of configurations based on the difference between their energy in a reference state and another state.
Class MBAREstimator:
- Uses the MBAR method to reweight samples and estimate free energies.
- Can add/remove samples and states, compute energy matrices, and optimize weights using MBAR.
- Also contains functions to compute covariance and estimate effective sample size.
Here we would tell you how to create a MBAR Estimator and use it.
- Initialization: Create an instance of the
MBAREstimator:
estimator = MBAREstimator()-
Prepare the Sampling State and Samples:
-
Define the name of the state:
state_name = "prm"- Create a state using
OpenMMSampleState. This state is defined by certain parameters including an XML file presumably containing the force field parameters, a PDB file with molecular configurations, and other physical conditions:
state = OpenMMSampleState(state_name, "prm.xml", "box.pdb", temperature=298.0, pressure=1.0)- Load a trajectory (sequence of molecular configurations) using
md.load(in mdtraj) and slice it to discard the initial configurations (the first 50 frames in this case):
traj = md.load("init.dcd", top="box.pdb")[50:]- Create a sample using the loaded trajectory and the previously defined state name:
sample = Sample(traj, state_name)-
Add the State and Sample to the Estimator:
-
Add the created state to the estimator:
estimator.add_state(state)- Add the sample to the estimator:
estimator.add_sample(sample)- Optimization: Invoke the
optimize_mbarfunction (which calls the external toolpymbarin the background) to estimate the partition functions for all sampled states.
estimator.optimize_mbar()From the provided steps, it's clear that the MBAREstimator works as follows:
-
Initialize the estimator. The variables that need to be initialized in MBAREstimator include two parts:
-
All sampling state information and samples it contains.
-
The sampling state partition function estimated based on the samples and sampling state information.
-
Prepare the states and the trajectory samples that represent these states.
-
Add the states and samples to the estimator.
-
Optimize the weights using the MBAR method to reweight the samples and estimate thermodynamic quantities.
Note: The operations are not involved in differentiation and do not depend on jax. Once the estimator is initialized, it can directly provide $W(x_{n})$in a differentiable manner.