HUMAnN Workshop on Genomics 2023
HUMAnN (the HMP Unified Metabolic Analysis Network) is a method for efficiently and accurately profiling the abundance of microbial metabolic pathways and other molecular functions from metagenomic or metatranscriptomic sequencing data. It is appropriate for any type of microbial community, not just the human microbiome (the name "HUMAnN" is a historical product of the method's origins in the Human Microbiome Project).
HUMAnN 3.0 can be installed 1) from PyPI, 2) as source code from the HUMAnN Github repository, or 3) using our quickstart documentation.
Support for HUMAnN is available via the HUMAnN channel of the bioBakery Support Forum.
- 1. Setup
- 2. Metagenome functional profiling
- 3. Manipulating HUMAnN output tables
- 4. Advanced HUMAnN topics
- 5. Plotting stratified functions
NOTE: Bowtie2, DIAMOND, and MinPath are automatically installed when installing HUMAnN. If these dependencies do not appear to be installed after installing HUMAnN with pip, it might be that your environment is setup to use wheels instead of installing from source. HUMAnN must be installed from source for it to also be able to install dependencies. To force pip to install HUMAnN from source, add one of the following options to your install command, --no-use-wheel or --no-binary :all:.
NOTE: If you are using a bioBakery machine image (e.g. in Google Cloud) you do not need to install HUMAnN because the software and its dependencies are already available.
The easiest way to install HUMAnN is with pip, Conda, or Docker. For other options, please refer to the HUMAnN User Manual.
To install with pip:
pip install humann
You can also install via Conda, which will additionally install MetaPhlAn and utility dependencies. Before doing so, make sure to configure your channels so that biobakery is at the top of the list:
conda install -c biobakery humann
By default, a pip (or other) HUMAnN install does not include the full-scale nucleotide and protein sequence databases needed for real-world metagenome or metatranscriptome profiling, as these are quite large (several GBs). The humann_databases script can be used to fetch these from the web and connect them to your HUMAnN installation. For tutorial purposes, we'll be using (small) demo databases, which are also fetchable with humann_databases (if not already available).
humann_databases --download chocophlan DEMO humann_dbs
humann_databases --download uniref DEMO_diamond humann_dbs
The commands above download the demo-scale databases to a folder named humann_dbs in the current working directory. Please note that you can download these (and full-scale) databases to any location on your machine: HUMAnN will track where you have placed the databases and update its configuration file.
After installation from pip or another source, you may optionally test your local HUMAnN environment (this takes ~1 minute):
humann_test
Which yields (highly abbreviated):
test_calculate_percent_identity (basic_tests_nucleotide_search.TestBasicHumann2NucleotideSearchFunctions) ... ok
test_calculate_percent_identity_insert_delete (basic_tests_nucleotide_search.TestBasicHumann2NucleotideSearchFunctions) ... ok
test_calculate_percent_identity_missing_cigar_identifier (basic_tests_nucleotide_search.TestBasicHumann2NucleotideSearchFunctions) ... ok
test_calculate_percent_identity_missing_md_field (basic_tests_nucleotide_search.TestBasicHumann2NucleotideSearchFunctions) ... ok
test_calculate_percent_identity_multiple_M_cigar_fields (basic_tests_nucleotide_search.TestBasicHumann2NucleotideSearchFunctions) ... ok
...
HUMAnN provides organism-specific community functional profiles, and to do so it first detects the organisms present in a community using MetaPhlAn. As MetaPhlAn is thus a prerequisite for running the HUMAnN software, please ensure that you've installed MetaPhlAn before running HUMAnN. You can test if MetaPhlAn is available in your system path by executing metaphlan --version.
A note on inspecting files from the command line: During this tutorial you'll inspect many HUMAnN input and output files, many of which are tab-delimitted tables (TSV files). For simplicity the tutorial uses less -S file.tsv for this task. To match the more spreadsheet-like columns seen in the tutorial document, you would first use the column command, as in:
column -t -s $'\t' file.tsv | less -S
HUMAnN can start from a few different types of input data each in a few different types of formats:
- Quality-controlled shotgun sequencing reads
- This is the most common starting point
- A metagenome (DNA reads) or metatranscriptome (RNA reads)
- Formats:
fastq,fastq.gz,fasta, orfasta.gz
- Pre-computed mappings of reads to database sequences
- Formats:
sam,bam, orblastm8
- Formats:
- Pre-computed (typically gene) abundance tables
- Formats:
tsvorbiom
- Formats:
This tutorial will use demo input files distributed with the HUMAnN installation under the examples directory. If you installed with pip or conda and don't have these handy, you can obtain copies by right-clicking these links and selecting "save link as":
If you are running this tutorial as part of a short course, all input files have been pre-downloaded into the cloud instances. To run this tutorial from the correct location, enter:
cd ~/workshop_materials/humann/HUMAnN_tutorial
A canonical HUMAnN run uses nucleotide mapping and translated search to provide organism-specific gene and pathway abundance profiles from a single metagenome. By default, gene families are annotated using UniRef90 definitions and pathways using MetaCyc definitions. From the examples folder (or the location where you saved demo.fastq.gz above) execute the following:
conda activate humann36
humann --input demo.fastq.gz --output demo_fastq
(Note1: If you have more than 1 CPU available for computation, you can use N of them by adding --threads N to the above command.)
This command yields (blank lines removed):
Creating output directory: demo_fastq
Output files will be written to: demo_fastq
Running metaphlan ........
Found g__Bacteroides.s__Bacteroides_dorei : 57.96% of mapped reads
Found g__Bacteroides.s__Bacteroides_vulgatus : 42.04% of mapped reads
Total species selected from prescreen: 2
Selected species explain 100.00% of predicted community composition
Creating custom ChocoPhlAn database ........
Running bowtie2-build ........
Running bowtie2 ........
Total bugs from nucleotide alignment: 2
g__Bacteroides.s__Bacteroides_vulgatus: 1195 hits
g__Bacteroides.s__Bacteroides_dorei: 1260 hits
Total gene families from nucleotide alignment: 545
Unaligned reads after nucleotide alignment: 88.3095238095 %
Running diamond ........
Aligning to reference database: uniref90_demo_prots_v201901b.dmnd
Total bugs after translated alignment: 3
g__Bacteroides.s__Bacteroides_vulgatus: 1195 hits
g__Bacteroides.s__Bacteroides_dorei: 1260 hits
unclassified: 252 hits
Total gene families after translated alignment: 559
Unaligned reads after translated alignment: 87.2000000000 %
Computing gene families ...
Computing pathways abundance and coverage ...
Output files created:
demo_fastq/demo_genefamilies.tsv
demo_fastq/demo_pathabundance.tsv
demo_fastq/demo_pathcoverage.tsv
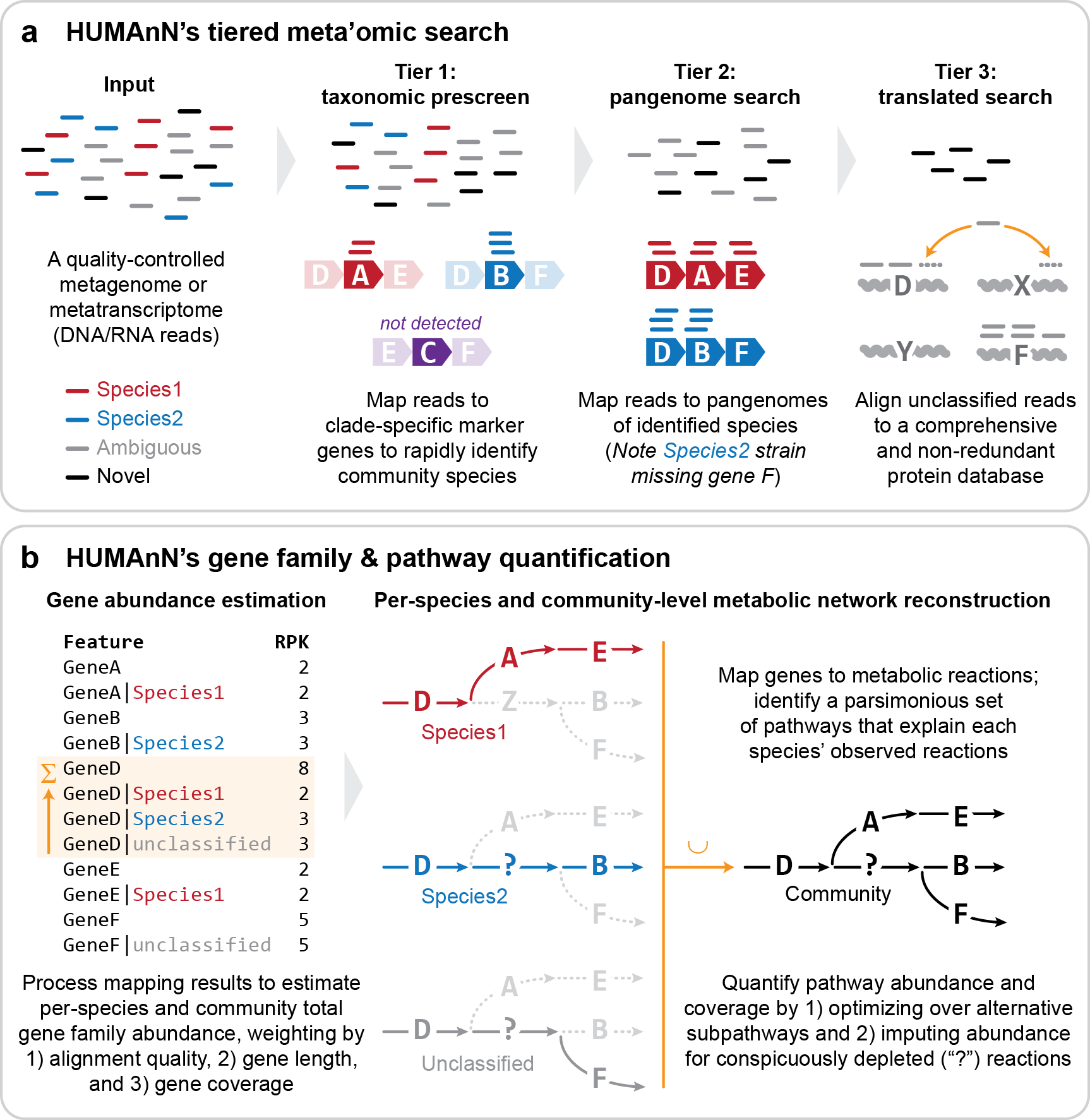
Note the three phases of HUMAnN's tiered search in the above log: 1) identifying community species (with MetaPhlAn), 2) mapping reads to community pangenomes (with bowtie2), and 3) aligning unmapped reads to a protein database (with DIAMOND).
- Why is nucleotide-level search generally faster than translated search?
- What makes nucleotide-level search extra fast in HUMAnN?
(Note: The outputs in this tutorial were generated using HUMAnN 3.0.0 with its corresponding demo databases. If you are running a different version of HUMAnN or are using a different set of databases, the outputs might differ.)
When HUMAnN is run from any input type, three main output files will be created:
$OUTPUT_DIR/$SAMPLE_genefamilies.tsv
$OUTPUT_DIR/$SAMPLE_pathabundance.tsv
$OUTPUT_DIR/$SAMPLE_pathcoverage.tsv
For the demo.fastq.gz file analyzed above, these look like:
demo_fastq/demo_genefamilies.tsv
demo_fastq/demo_pathabundance.tsv
demo_fastq/demo_pathcoverage.tsv
Let's inspect the contents of the individual files:
This file contains the abundances of each gene family in the community in reads per kilobase (RPK) units. Each gene family's total abundance is broken down into the contributions from individual organisms when possible (delineated by the pipe character). For example:
UniRef90_G1UL42|g__Bacteroides.s__Bacteroides_dorei
Read mappings generated during (organism-agnostic) translated search are assigned to an unclassified bin. We refer to this format as "stratified output" and the individual per-organism and unclassified contributions are referred to as "stratifications". By default, gene families are identified by their unique UniRef90 identifiers (e.g. UniRef90_G1UL42 in the example above). While these are convenient for computing, they are not very easy to interpret by eye. We'll see how to attach human-readable names to UniRef90 (and other) identifiers in a subsequent section.
You can also learn more about a given UniRef family by looking up its representative protein on UniProt thusly (note the removal of the UniRef90_ prefix):
https://www.uniprot.org/uniprot/G1UL42
Reads that map to gene families that lack UniRef90 identifiers are represented as UniRef90_unknown while unmapped reads are collected as UNMAPPED. The total coverage depth (RPK) of the sample is thus represented by summing either 1) UNMAPPED, UniRef90_unknown, and all UniRef IDs prior to organism-specific decomposition, or 2) UNMAPPED, UniRef90_unknown|<organism>, and all other UniRef_*|<organism> IDs. The sum of either set of values can then be normalized to CPMs (Copies Per Million) or relative abundances.
To inspect the demo_genefamilies.tsv file, use:
column -t -s $'\t' demo_fastq/demo_genefamilies.tsv | less -S
Which yields:
# Gene Family demo_Abundance-RPKs
UNMAPPED 18312.0000000000
UniRef90_G1UL42 333.3333333333
UniRef90_G1UL42|g__Bacteroides.s__Bacteroides_dorei 333.3333333333
UniRef90_I9QXW8 333.3333333333
UniRef90_I9QXW8|g__Bacteroides.s__Bacteroides_dorei 333.3333333333
UniRef90_A0A174QBF2 200.0000000000
UniRef90_A0A174QBF2|g__Bacteroides.s__Bacteroides_vulgatus 200.0000000000
UniRef90_A0A078RDY6 166.6666666667
UniRef90_A0A078RDY6|g__Bacteroides.s__Bacteroides_vulgatus 166.6666666667
UniRef90_G1ULL9 166.6666666667
UniRef90_G1ULL9|g__Bacteroides.s__Bacteroides_vulgatus 166.6666666667
UniRef90_A0A015QIN1 66.6666666667
UniRef90_A0A015QIN1|g__Bacteroides.s__Bacteroides_vulgatus 66.6666666667
Let's find some of the "unclassified" stratifications in this file using the grep command:
grep unclassified demo_fastq/demo_genefamilies.tsv | less -S
Which yields:
UniRef90_Q9ZUH4|unclassified 18.3746362785
UniRef90_W8YTG4|unclassified 13.9024544125
UniRef90_Q6XMI3|unclassified 13.5681161633
UniRef90_U5FT06|unclassified 13.4157878980
UniRef90_Z9K4C5|unclassified 12.7418650404
UniRef90_U3KI10|unclassified 12.3842423992
UniRef90_B8RBV1|unclassified 12.1442525829
UniRef90_U3KP22|unclassified 11.6963164253
UniRef90_Z9JLB1|unclassified 11.5045104368
UniRef90_Q8GT21|unclassified 11.2241858840
- HUMAnN can quantify protein families at 90 or 50% identity (via mapping to UniRef90 and UniRef50 representatives, respectively). What are the advantages of quantifying UniRef90 gene families?
- UniRef50 families?
This file lists the abundances of each pathway in the community, also in RPK units as described for gene families. For details on the calculation of pathway abundances from constituent gene family abundances, see the HUMAnN User Manual. Like gene families, pathways are broken down into per-organism contributions when possible (again delineated by the | character), with the total read abundance consisting of pathways assigned to organisms, UNMAPPED reads, and UNINTEGRATED reads that are attributable to specific gene families (typically also within specific organisms), but for gene families that are not annotated to any pathway in the corresponding database (e.g. MetaCyc).
As with gene families, pathway RPKs can be normalized to copies per million or relative abundances by summing either 1) UNMAPPED reads, UNINTEGRATED organism-specific features, and organism-specific (i.e. pipe-containing) pathways, or 2) UNMAPPED reads, total UNINTEGRATED features, and pathways that are not broken down per organism.
To inspect the demo_pathabundance.tsv file, use:
column -t -s $'\t' demo_fastq/demo_pathabundance.tsv | less -S
Which yields:
# Pathway demo_Abundance
UNMAPPED 9520.1365299060
UNINTEGRATED 4236.5362970855
UNINTEGRATED|unclassified 54.1292645042
PWY-5423: oleoresin monoterpene volatiles biosynthesis 18.3746362785
PWY-5423: oleoresin monoterpene volatiles biosynthesis|unclassified 18.3746362785
PWY-4203: volatile benzenoid biosynthesis I (ester formation) 13.3772872602
PWY-4203: volatile benzenoid biosynthesis I (ester formation)|unclassified 13.3772872602
(Note to instructors: Pathway coverage output is being revised; you may skip reviewing it during tutorials pending these updates.)
- Unlike gene family abundance, stratified pathway abundance does not necessarily sum to yield the community total pathway abundance. Why not? (Hint: HUMAnN estimates the number of complete pathways in the community or a given stratification.)
The raw results of mapping metagenome/metatranscriptome reads against a gene family nucleotide database can be saved as a SAM/BAM file and used to run HUMAnN after the mapping (e.g. bowtie2) step. The only requirement is that gene family names provided in such a file be annotated using either 1) identifiers compatible with HUMAnN's default mapping to UniRef, or 2) UniRef identifiers directly. Unmapped reads in such an input file are searched against a non-organism-specific protein database as are unmapped reads in a "full" HUMAnN run. Inspect the demo sam file:
less -S demo.sam
We can transpose and view a single row (per-read alignment) as followed:
head -1 demo.sam | tr "\t" "\n"
The head -1 command extracts the first line of the file, while the tr command replaces tabs with newlines:
s__Bacteroides_dorei_read000001
16
357276|g__Bacteroides.s__Bacteroides_dorei|UniRef90_R7NX76|UniRef50_R5GDB8|927
423
11
100M
*
0
0
TTTCGTATTAGTGGAGTATGGCATTGAAAGTACGGATGATGACACGTTGCGCAGGATAAACCGGGGACATACTTTTGCCGTTTCTGCAGAGGCTGTTCGG
IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII
AS:i:-12
XS:i:-30
XN:i:0
XM:i:2
XO:i:0
XG:i:0
NM:i:2
MD:Z:3T38A57
YT:Z:UU
We can provide this sam file as input to HUMAnN using a command similar to the one used above for the fastq file:
humann --input demo.sam --output demo_sam
which yields (blank lines removed):
Creating output directory: demo_sam
Output files will be written to: demo_sam
Process the sam mapping results ...
Computing gene families ...
Computing pathways abundance and coverage ...
Output files created:
demo_sam/demo_genefamilies.tsv
demo_sam/demo_pathabundance.tsv
demo_sam/demo_pathcoverage.tsv
Note the creation of the three expected output files.
- Gap filling allows us to quantify a pathway even when a small number of its reactions are conspicuously absent. What are some potential pros and cons of this approach in real data?
HUMAnN typically searches metagenome/metatranscriptome reads against two databases: first a nucleotide database using rapid mapping, then a broader protein database using more computationally expensive translated search. One can execute HUMAnN using either search alone, however; using nucleotides only is more computationally efficient but less sensitive, and using translated search alone can emulate HUMAnN 1.0 output or be carried out even for very novel or unusual microbial communities (i.e. those with few reference genomes available).
If BLAST tab-delimited M8 format search results have already been generated for HUMAnN-compatible protein IDs (UniRef90 by default), they can be run by providing the appropriate input format. Let's examine such a file:
less -S demo.m8
Which yields:
s__Bacteroides_vulgatus_read000635|100 UniRef90_X8JRH1|978 57.9 19 8 0 95 39 189 207 2.0e-01 21.6
unclassified_read000001|100 UniRef90_W8YTG4|1614 90.6 32 3 0 98 3 382 413 9.0e-15 65.9
unclassified_read000002|100 UniRef90_U5FT06|1224 90.9 33 3 0 100 2 127 159 2.1e-14 64.7
unclassified_read000003|100 UniRef90_V5V2L3|2169 90.6 32 3 0 3 98 388 419 4.5e-14 63.5
unclassified_read000004|100 UniRef90_U5FT06|1224 90.6 32 3 0 99 4 148 179 4.6e-14 63.5
unclassified_read000005|100 UniRef90_X8FHU5|1080 90.6 32 3 0 98 3 240 271 3.1e-15 67.4
unclassified_read000006|100 UniRef90_Z9JXD8|1428 90.6 32 3 0 98 3 22 53 2.6e-14 64.3
unclassified_read000007|100 UniRef90_Q6XMI3|1137 90.6 32 3 0 3 98 155 186 2.0e-14 64.7
unclassified_read000007|100 UniRef90_Q9SPV4|1077 65.6 32 11 0 3 98 129 160 1.5e-09 48.5
unclassified_read000008|100 UniRef90_Z9JUT2|1308 90.6 32 3 0 98 3 222 253 2.4e-15 67.8
unclassified_read000009|100 UniRef90_X5M7Z0|1134 93.9 33 2 0 1 99 202 234 2.9e-16 70.9
To run HUMAnN using this file as input, use:
humann --input demo.m8 --output demo_m8
This again produces the three expected outputs (blank lines removed):
Creating output directory: /demo_m8
Output files will be written to: /demo_m8
Process the blastm8 mapping results ...
Computing gene families ...
Computing pathways abundance and coverage ...
Output files created:
/demo_m8/demo_genefamilies.tsv
/demo_m8/demo_pathabundance.tsv
/demo_m8/demo_pathcoverage.tsv
- The BLAST m8 output fields allow us to compute the coverage of the database sequence by a single sequencing read (subject coverage) and the coverage of the read by the database sequence (query coverage). Why is subject coverage almost always smaller than query coverage?
Conversely, to run HUMAnN without any translated protein search (i.e. using rapid nucleotide mapping alone), one can provide the --bypass-translated-search flag. This applies to FASTA, FASTQ, SAM/BAM, or other raw nucleotide inputs:
humann --input demo.fastq.gz --output demo_fastq_fast --bypass-translated-search
Note the absence of a diamond (translated search) step in this run compared to the demo_fastq analysis above:
Creating output directory: demo_fastq_fast
Output files will be written to: demo_fastq_fast
Running metaphlan ........
Found g__Bacteroides.s__Bacteroides_dorei : 58.10% of mapped reads
Found g__Bacteroides.s__Bacteroides_vulgatus : 41.90% of mapped reads
Total species selected from prescreen: 2
Selected species explain 100.00% of predicted community composition
Creating custom ChocoPhlAn database ........
Running bowtie2-build ........
Running bowtie2 ........
Total bugs from nucleotide alignment: 2
g__Bacteroides.s__Bacteroides_vulgatus: 1195 hits
g__Bacteroides.s__Bacteroides_dorei: 1260 hits
Total gene families from nucleotide alignment: 545
Unaligned reads after nucleotide alignment: 88.3095238095 %
Bypass translated search
Computing gene families ...
Computing pathways abundance and coverage ...
Output files created:
demo_fastq_fast/demo_genefamilies.tsv
demo_fastq_fast/demo_pathabundance.tsv
demo_fastq_fast/demo_pathcoverage.tsv
It is informative to compare the results of this demo file with and without translated search; even in a small, subsampled input, including amino acid search is more computationally costly but recovers additional gene families. To count unique gene families in the original file, execute:
cut -f1 demo_fastq/demo_genefamilies.tsv | tail -n +3 | grep -v "|" | sort -u | wc -l
What is this command doing?
cut -f1extracts the first column of the file, i.e. gene identifiers;tail -n +3removes the first two lines of the file, i.e. the header andUNMAPPED;grep -v "|"removes the stratified headers;sort -uisolates the unique headers;wc -lcounts those headers.
The resulting answer is 559 (unique gene families). The same command executed on the file demo_fastq_fast/demo_genefamilies.tsv yields 545 unique gene families. We infer that 559 - 545 = 14 additional gene families were uncovered using translated search. (On a non-demo metagenome, the relative number of additional gene families uncovered by translated search would be far larger!)
In general, any HUMAnN intermediate file can be saved and used to resume processing from an intermediate point in the analysis pipeline, and several steps including taxonomic pre-screening, nucleotide search, or amino acid search can be independently skipped. For more information, see the relevant sections of the HUMAnN User Manual.
- Under what circumstances would skipping translated search be reasonable?
- Under what circumstances would skipping translated search be a bad idea?
To facilitate comparisons between samples with different sequencing depths, it is important to normalize HUMAnN's default RPK values prior to performing statistical analyses. Sum-normalization to relative abundance or "copies per million" (CPM) units are common approaches for this; these methods are implemented in the humann_renorm_table script (with special considerations for HUMAnN's output formats, e.g. feature stratification). Let's normalize our gene abundances to CPM units:
humann_renorm_table --input demo_fastq/demo_genefamilies.tsv \
--output demo_fastq/demo_genefamilies-cpm.tsv --units cpm --update-snames
Inspect the output file demo_fastq/demo_genefamilies-cpm.tsv using the less command as we did above:
# Gene Family demo_Abundance-CPM
UNMAPPED 690444
UniRef90_G1UL42 12568.2
UniRef90_G1UL42|g__Bacteroides.s__Bacteroides_dorei 12568.2
UniRef90_I9QXW8 12568.2
UniRef90_I9QXW8|g__Bacteroides.s__Bacteroides_dorei 12568.2
UniRef90_A0A174QBF2 7540.89
UniRef90_A0A174QBF2|g__Bacteroides.s__Bacteroides_vulgatus 7540.89
UniRef90_A0A078RDY6 6284.08
UniRef90_A0A078RDY6|g__Bacteroides.s__Bacteroides_vulgatus 6284.08
UniRef90_G1ULL9 6284.08
UniRef90_G1ULL9|g__Bacteroides.s__Bacteroides_vulgatus 6284.08
UniRef90_A0A015QIN1 2513.63
UniRef90_A0A015QIN1|g__Bacteroides.s__Bacteroides_vulgatus 2513.63
Note the difference in scale: units tend to be larger than in the original file as a result of being scaled to sum to 1 million. Also note that the sample header has been updated from demo_Abundance-RPK to demo_Abundance-CPM courtesy of the --update-snames flag.
By default, humann_renorm_table 1) forces the community total abundances to sum to 1 million and then 2) adjusts the stratifications so that they continue to behave as fractions of community total abundances (see humann_renorm_table -h for additional options). Let's verify that the unstratified abundance values sum up to 1 million:
grep -v "#" demo_fastq/demo_genefamilies-cpm.tsv | grep -v "|" | cut -f2 | paste -s -d+ - | bc
What is this command doing?
grep -v "#": excludes lines containing "#", in this case the file's header;grep -v "|": excludes lines containing "|", which mark stratified abundances;cut -f2: extracts the second column of the file containing the abundances;paste -s -d+ -: converts the column of numbers to a summation (1 + 2 + 3 + ...);bc: sums the numbers.
The final answer should be very close to 1 million (it may be slightly off due to rounding error). A small modification of the above command can be used to find the sum of the stratified values (this sum will be < 1,000,000 unless you also account for the UNMAPPED value).
- HUMAnN natively normalizes gene family abundance by gene length. Why is this step important? (In other words, why are RPK units more interpretable than raw read counts?)
HUMAnN's default "units" of microbial function are UniRef gene families (which we use internally to compute reaction abundances, and from there pathway abundances). However, starting from gene family abundance, it is possible to reconstruct the abundance of other functional categories in a microbiome using the humann_regroup_table script. Inspect the regrouping options using the humann_regroup_table -h command.
Let's regroup our CPM-normalized gene family abundance values to MetaCyc reaction (RXN) abundances, which are bundled with the default HUMAnN installation:
humann_regroup_table --input demo_fastq/demo_genefamilies-cpm.tsv \
--output demo_fastq/rxn-cpm.tsv --groups uniref90_rxn
We are given the output message:
Original Feature Count: 560; Grouped 1+ times: 60 (10.7%); Grouped 2+ times: 13 (2.3%)
Indicating that the original file contained 560 features (UniRef90 gene families and UNMAPPED). Of these, 60 were annotated to a RXN, and 13 were annotated to more than one RXN. Inspect the output file demo_fastq/rxn-cpm.tsv using the less command:
# Gene Family demo_Abundance-CPM
UNMAPPED 690444.0
UNGROUPED 290292.34000000026
UNGROUPED|g__Bacteroides.s__Bacteroides_dorei 139621.93409999993
UNGROUPED|g__Bacteroides.s__Bacteroides_vulgatus 148318.30399999986
UNGROUPED|unclassified 2352.097
2.5.1.19-RXN 233.716
2.5.1.19-RXN|g__Bacteroides.s__Bacteroides_dorei 133.704
2.5.1.19-RXN|g__Bacteroides.s__Bacteroides_vulgatus 100.012
2.7.13.3-RXN 236.729
2.7.13.3-RXN|g__Bacteroides.s__Bacteroides_dorei 172.166
2.7.13.3-RXN|g__Bacteroides.s__Bacteroides_vulgatus 64.5624
2.7.7.13-RXN 398.989
2.7.7.13-RXN|g__Bacteroides.s__Bacteroides_dorei 119.697
2.7.7.13-RXN|g__Bacteroides.s__Bacteroides_vulgatus 279.292
We see that UniRef90 features have been regrouped as RXN features, such as 2.5.1.19-RXN. A new stratified feature also appears in the output: UNGROUPED. This feature reports the total abundance of features that were not otherwise included in a group (similar to the UNINTEGRATED feature of the pathways output). If each UniRef90 gene family were regrouped into at most one RXN, the total community abundance of features in the regrouped file (including UNGROUPED and UNMAPPED) would still be 1 million.
The actual sum here (1,024,360) is slightly higher than 1 million due to the 13 UniRef90 features whose abundances were counted in two (or more) different RXN features. You can verify this by modifying the summation command we used above:
grep -v "#" demo_fastq/rxn-cpm.tsv | grep -v "|" | cut -f2 | paste -s -d+ - | bc
You could force the RXN abundances themselves to sum to 1 million by running humann_renorm_table on the above table, or by regrouping UniRef90s to RXNs before renormalizing their abundances (and then normalizing at the RXN stage). The results will be the same! (That said, in practice, we typically normalize only once at the gene family level to adjust for initial differences in sample sequencing depth.)
- When regrouping genes to RXNs (or ECs or KOs), we sum over the gene sequences belonging to a particular group. Under what circumstance would it make sense to average over the members of a group rather than sum? (Hint: the answer is related to the difference between the "is_a" and "part_of" relationships used in the Gene Ontology.)
So far we've been looking at a bunch of UniRef90 codes and RXN codes. While HUMAnN is happy to work with these all day, it's typically helpful to attach some human-readable descriptions of these IDs to facilitate biological interpretation. This can be accomplished with the humann_rename_table script. Let's attach names to the RXN table we generated above:
humann_rename_table --input demo_fastq/rxn-cpm.tsv \
--output demo_fastq/rxn-cpm-named.tsv --names metacyc-rxn
We are told that the script Renamed 116 of 118 entries (98.31%), suggesting that most features were successfully renamed (indeed, the two features that did not receive new names were the special UNMAPPED and UNGROUPED features). Let's confirm this by inspecting the new output table with less:
# Gene Family demo_Abundance-CPM
UNMAPPED 690444.0
UNGROUPED 290292.34000000026
UNGROUPED|g__Bacteroides.s__Bacteroides_dorei 139621.93409999993
UNGROUPED|g__Bacteroides.s__Bacteroides_vulgatus 148318.30399999986
UNGROUPED|unclassified 2352.097
2.5.1.19-RXN: (expasy) 3-phosphoshikimate 1-carboxyvinyltransferase [2.5.1.19] 233.716
2.5.1.19-RXN: (expasy) 3-phosphoshikimate 1-carboxyvinyltransferase [2.5.1.19]|g__Bacteroides.s__Bacteroides_dorei 133.704
2.5.1.19-RXN: (expasy) 3-phosphoshikimate 1-carboxyvinyltransferase [2.5.1.19]|g__Bacteroides.s__Bacteroides_vulgatus 100.012
2.7.13.3-RXN: (expasy) Histidine kinase [2.7.13.3] 236.729
2.7.13.3-RXN: (expasy) Histidine kinase [2.7.13.3]|g__Bacteroides.s__Bacteroides_dorei 172.166
2.7.13.3-RXN: (expasy) Histidine kinase [2.7.13.3]|g__Bacteroides.s__Bacteroides_vulgatus 64.5624
2.7.7.13-RXN: (expasy) Mannose-1-phosphate guanylyltransferase [2.7.7.13] 398.989
2.7.7.13-RXN: (expasy) Mannose-1-phosphate guanylyltransferase [2.7.7.13]|g__Bacteroides.s__Bacteroides_dorei 119.697
2.7.7.13-RXN: (expasy) Mannose-1-phosphate guanylyltransferase [2.7.7.13]|g__Bacteroides.s__Bacteroides_vulgatus 279.292
It turns out that our friend 2.5.1.19-RXN is actually 3-phosphoshikimate 1-carboxyvinyltransferase, though you probably already knew that... In the event that a name can't be found for a non-special feature (e.g. UNMAPPED), the script will assign it the name NO_NAME for consistency.
The regrouping and renaming options used above are limited for demo purposes. Just as can install full-scale nucleotide and protein databases for real-world HUMAnN use, you can install a full-scale "utility mapping" database to help out with your downstream analyses. See the humann_databases script for acquiring these databases.
- From a statistical testing perspective, what could be an advantage of regrouping raw gene abundance to a smaller list of gene groups?
- What is a more practical advantage of regrouping?
It is in some cases useful to understand the collection of intermediate files generated by the individual steps of a typical HUMAnN run:
ls -lh demo_fastq/demo_humann_temp/
Which yields:
-rw-rw-r-- 1 15M May 20 14:26 demo_bowtie2_aligned.sam
-rw-rw-r-- 1 3.9M May 20 14:26 demo_bowtie2_aligned.tsv
-rw-rw-r-- 1 17M May 20 14:26 demo_bowtie2_index.1.bt2
-rw-rw-r-- 1 5.4M May 20 14:26 demo_bowtie2_index.2.bt2
-rw-rw-r-- 1 225K May 20 14:26 demo_bowtie2_index.3.bt2
-rw-rw-r-- 1 5.4M May 20 14:26 demo_bowtie2_index.4.bt2
-rw-rw-r-- 1 17M May 20 14:26 demo_bowtie2_index.rev.1.bt2
-rw-rw-r-- 1 5.4M May 20 14:26 demo_bowtie2_index.rev.2.bt2
-rw-rw-r-- 1 2.5M May 20 14:26 demo_bowtie2_unaligned.fa
-rw-rw-r-- 1 27M May 20 14:26 demo_custom_chocophlan_database.ffn
-rw-rw-r-- 1 88K May 20 14:26 demo_diamond_aligned.tsv
-rw-rw-r-- 1 2.4M May 20 14:26 demo_diamond_unaligned.fa
-rw-rw-r-- 1 24K May 20 14:27 demo.log
-rw-rw-r-- 1 19K May 20 14:25 demo_metaphlan_bowtie2.txt
-rw-rw-r-- 1 2.3K May 20 14:26 demo_metaphlan_bugs_list.tsv
These include SAM files containing the alignment of input reads against the default nucleotide database, which are first reduced by counting up and tabulating the hits to each annotated gene family. It can be useful to reprocess either the reads unaligned after nucleotide mapping (bowtie2_unaligned.fa) or, more likely, the reads still unaligned after amino acid search. Finally, the MetaPhlAn2 bowtie2.txt intermediate output is itself saved. For more information, see the HUMAnN User Manual's output file section.
It is typical to execute HUMAnN on multiple individual samples, then merge the resulting gene families or pathways into a single tab-delimited multi-sample table. When combining or comparing any HUMAnN output, it is critical to normalize the default RPK abundance units (which will be sensitive to the sequencing depth of each sample) into CPM units (which will not be).
We can demonstrate this by downloading a collection of six subsampled input files from the Human Microbiome Project. The original files, and many others, can be downloaded from the HMP DACC.
- 763577454-SRS014459-Stool.fasta
- 763577454-SRS014464-Anterior_nares.fasta
- 763577454-SRS014470-Tongue_dorsum.fasta
- 763577454-SRS014472-Buccal_mucosa.fasta
- 763577454-SRS014476-Supragingival_plaque.fasta
- 763577454-SRS014494-Posterior_fornix.fasta
Click on each individual subsampled file to download. Open a terminal and change directories into the folder where you have downloaded the files.
You will see something like the following contents if you run ls -lh:
-rw-r--r-- 1 2.7M Jun 4 11:33 763577454-SRS014459-Stool.fasta
-rw-r--r-- 1 2.6M Jun 4 11:33 763577454-SRS014464-Anterior_nares.fasta
-rw-r--r-- 1 2.7M Jun 4 11:33 763577454-SRS014470-Tongue_dorsum.fasta
-rw-r--r-- 1 2.7M Jun 4 11:34 763577454-SRS014472-Buccal_mucosa.fasta
-rw-r--r-- 1 2.7M Jun 4 11:34 763577454-SRS014476-Supragingival_plaque.fasta
-rw-r--r-- 1 2.7M Jun 4 11:34 763577454-SRS014494-Posterior_fornix.fasta
Process each input file into a single output directory:
humann -i 763577454-SRS014459-Stool.fasta -o hmp_subset
humann -i 763577454-SRS014464-Anterior_nares.fasta -o hmp_subset
humann -i 763577454-SRS014470-Tongue_dorsum.fasta -o hmp_subset
humann -i 763577454-SRS014472-Buccal_mucosa.fasta -o hmp_subset
humann -i 763577454-SRS014476-Supragingival_plaque.fasta -o hmp_subset
humann -i 763577454-SRS014494-Posterior_fornix.fasta -o hmp_subset
Alternatively, if you're comfortable with shell scripting, you can use the following command to process all six at once:
for f in *.fasta; do humann -i $f -o hmp_subset; done
Regardless of how you run HUMAnN, this will result in six output directories each containing the three main analysis types (gene families, pathway abundances, and pathway coverages) along with intermediate files. Let's merge the 6 per-sample gene family abundance outputs into a single table for this dataset using the script
humann_join_tables:
humann_join_tables -i hmp_subset -o hmp_subset_genefamilies.tsv --file_name genefamilies
Unfortunately the resulting file is not very interesting: because we're working with small demo databases, we don't identify much of anything as known from these subsampled HMP metagenomes. The same would not be true if we were using full-scale HUMAnN databases, which recruit the vast majority of reads from metagenomes of the human microbiome. Note also that, in a real-world application, it would be particularly important to normalize our merged file to adjust for differences in sequencing depth across the samples.
humann_renorm_table -i hmp_subset_genefamilies.tsv -o hmp_subset_genefamilies-cpm.tsv --units cpm
For more information, see the HUMAnN User Manual's description of a standard analysis workflow.
- When analyzing less-well-characterized metagenomes, we recommend using a UniRef50-based HUMAnN workflow rather than the default UniRef90-based workflow. Why might that be the case, and how would you expect mapping rates in HUMAnN's three search tiers to change in that context?
For this section, we'll operate on a table of pathway abundances from ~400 samples from the Human Microbiome Project (as originally profiled by HUMAnN 2.0):
Download or copy (if in VM: cp input/hmp_pathabund.pcl .) this file to your working directory and examine it using less:
less -S hmp_pathabund.pcl
Which yields:
FEATURE \ SAMPLE SRS011084 SRS011086 SRS011090
STSite Stool Tongue_dorsum Buccal_mucosa
1CMET2-PWY: N10-formyl-tetrahydrofolate biosynthesis 0.000498359 0.000628096 0.000304951
1CMET2-PWY: N10-formyl-tetrahydrofolate biosynthesis|g__Acidovorax.s__Acidovorax_ebreus 0 0 0
1CMET2-PWY: N10-formyl-tetrahydrofolate biosynthesis|g__Bacteroides.s__Bacteroides_finegoldii 0 0 0
1CMET2-PWY: N10-formyl-tetrahydrofolate biosynthesis|g__Haemophilus.s__Haemophilus_influenzae 0 0 0
1CMET2-PWY: N10-formyl-tetrahydrofolate biosynthesis|g__Klebsiella.s__Klebsiella_pneumoniae 0 0 0
1CMET2-PWY: N10-formyl-tetrahydrofolate biosynthesis|g__Staphylococcus.s__Staphylococcus_epidermidis 0 0 0
1CMET2-PWY: N10-formyl-tetrahydrofolate biosynthesis|g__Veillonella.s__Veillonella_sp_oral_taxon_158 0 0 0
1CMET2-PWY: N10-formyl-tetrahydrofolate biosynthesis|unclassified 7.89502e-06 1.60101e-05 0
We can see three samples from three different body sites (via the STSite metadatum). A single pathway, N10-formyl-tetrahydrofolate biosynthesis (1CMET2-PWY), and its stratifications are visible. In these three samples, the pathway can only be assigned to the unclassified stratum, and with very low abundance. In a third sample (SRS011090) the pathway is only observed at the community level (how?).
Using MaAsLin 2.0 (or another method for making statistical associations with microbiome data), we can compare community total abundances from HUMAnN tables with sample metadata. In this case, a model relating community-total pathway abundance to sample body site reveals a strong enrichment for L-homoserine and L-methionine biosynthesis (METSYN-PWY) at the three oral sites (buccal mucosa, tongue, and plaque).
Now that we have identified this enrichment, use the grep command to explore the stratifications of this pathway from the hmp_pathabund.pcl file.
grep 'METSYN-PWY' hmp_pathabund.pcl | less -S
- Do the major contributing species seem reasonable for an orally-enriched pathway?
HUMAnN includes a utility humann_barplot to assist with visualizing stratified outputs. Let's use this tool to make a plot of the differentially abundant pathway we identified above:
humann_barplot --input hmp_pathabund.pcl --focal-metadata STSite --last-metadata STSite \
--output plot1.png --focal-feature METSYN-PWY
Use
see plot1.pngto open the resulting figure if working in a graphical environment.)

Not terribly informative... Let's try adding a "sort" on the summed stratified abundance:
humann_barplot --input hmp_pathabund.pcl --focal-metadata STSite --last-metadata STSite \
--output plot2.png --focal-feature METSYN-PWY --sort sum

A pattern has started to emerge: we can clearly see that the oral body sites are enriched on the left (high) end of the plot. We can continue this line of analysis by adding an additional sort on body site (metadata) and "logstack" scaling (i.e. scaling the tops of the stacked bars in proportion to the log of community abundance, while scaling species contributions linearly within total bar height):
humann_barplot --input hmp_pathabund.pcl --focal-metadata STSite --last-metadata STSite \
--output plot3.png --focal-feature METSYN-PWY --sort sum metadata --scaling logstack

We see now that the pathway is most strongly enriched in the buccal mucosa, where it is contributed almost exclusively by Streptococcus species. The pathway's relative abundance in the other two oral sites is generally lower (by about an order of magnitude, as emphasized on the log scale), and is more often "unclassified" at the tongue body site.
- How could you find other pathways with a similar abundance distribution in this functional profile?
- What might be causing the "unclassified" contributors of homoserine/methionine synthesis in tongue dorsum communities?
Let's examine one additional pathway, coenzyme A biosynthesis (COA-PWY), which is more broadly conserved across body sites:
humann_barplot --input hmp_pathabund.pcl --focal-metadata STSite --last-metadata STSite \
--output plot4.png --focal-feature COA-PWY --sort sum

Now sorting by total abundance is less informative. To remedy that, let's try some additional plotting options: 1) regrouping species to genera (--as-genera), 2) removing samples that lack the coenzyme A biosynthesis pathway (--remove-zeros), 3) and sorting on ecological similarity (--sort braycurtis):
humann_barplot --input hmp_pathabund.pcl --focal-metadata STSite --last-metadata STSite \
--output plot5.png --focal-feature COA-PWY --sort braycurtis --scaling logstack --as-genera --remove-zeros

Notably, sorting by the similarity of pathway contributions has tended to group samples from the same body site even without the explicit metadata sort. This is because, for broadly distributed pathways, organismal contributions tend to follow overall sample ecology (which tends to be quite different from one body site to the next). This plot also includes an appearance of the dark gray "other" bin, which in this case represents the sum of contributions from named genera that were outside of the top N we selected for coloring (default N=18).
- Is it surprising to see coenzyme A biosynthesis present in diverse human body site microbiomes?