forked from XSLiuLab/Workshop
-
Notifications
You must be signed in to change notification settings - Fork 0
/
Copy path几种常用的差异分析方法简介
342 lines (237 loc) · 12.7 KB
/
几种常用的差异分析方法简介
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
256
257
258
259
260
261
262
263
264
265
266
267
268
269
270
271
272
273
274
275
276
277
278
279
280
281
282
283
284
285
286
287
288
289
290
291
292
293
294
295
296
297
298
299
300
301
302
303
304
305
306
307
308
309
310
311
312
313
314
315
316
317
318
319
320
321
322
323
324
325
326
327
328
329
330
331
332
333
334
335
336
337
338
339
340
341
342
# 几种常用的差异分析方法简介
如今在生物学研究中,差异分析越来越普遍,也有许多做差异分析的方法可供选择。但是在实际应用中,大多数人不知道该使用哪种方法来处理自己的数据,所以今天我就来介绍下目前几种常用的差异分析方法及其适用场景。
**1.方差分析、T检验、卡方检验、秩和检验**
------

- 其实核心的区别在于:数据类型不一样。如果是定类和定类,此时应该使用卡方分析;如果是定类和定量,此时应该使用方差或者T检验。
- 方差和T检验的区别在于,对于T检验的X来讲,其只能为2个类别比如男和女。如果X为3个类别比如本科以下,本科,本科以上;此时只能使用方差分析。
**进一步细分**

#### **T检验**
------
t检验(student t检验)是应用t分布的特征,将t作为检验的统计量来进行统计推断方法。它对样本要求较小(例如n<30)。
主要用途:
- 样本均数与总体均数的差异比较
- 两样本均数的差异比较
**单样本t检验**
单样本t检验主要用于判断样本均数与总体均数是否存在显著差异。
**适用条件**
已知一个总体均数
已知一个样本均数及该样本标准差
样本正态分布或近似正态总体
实际应用中,当数据量足够大时,对样本正态分布要求不再严格。只要数据分布不是严重偏态,一般来说单样本t检验都是适用的。
R语言中可以用t.test函数进行t检验
从某小学六年级抽取10名学生,其身高(单位:cm),是否认为该学校六年级平均身高130cm?
~~~R
#生成数据
x <- c(130,120,130,110,130,135,129,124,130,134)
#t检验
t.test(x,mu = 130)
One Sample t-test
data: x
t = -1.1884, df = 9, p-value =
0.2651
alternative hypothesis: true mean is not equal to 130
95 percent confidence interval:
121.8702 132.5298
sample estimates:
mean of x
127.2
#结果显示,P=0.2651>0.05。在统计学上说明样本均数与总体均数没有差别。
~~~
**独立样本t检验**
独立样本t检验主要检验两个样本均数及其所代表的总体之间差异是否显著。
**适用条件**
- 独立性,各观察值之间相关独立
- 正态性,各样本均来自正态分布的总体
- 方差齐性,各样本所在总体的方差相等
方差齐性可以用car包leveneTest函数检验
leveneTest(y=,group =)
其中,y是两组样本组成的数据,group是两组样本的分组情况。
方差齐性检验之后,才可进行独立样本t检验。
用t.test(A,B,var.equal=TRUE,paired=FALSE)
A、B为数据集,var.equal=TRUE为方差齐性。paired=FALSE非配对样本。
示例:
(虚构)有两组学生(每组10人),一组采用传统教育,一组采用素质教育。一学期后,两组学生语文成绩(满分100)如下。问两组学生成绩之间差别是否显著。
~~~R
A <- c(85,84,95,73,77,65,85,93,90,91)
B <- c(87,96,77,80,79,96,93,82,84,86)
#方差齐性检验
#合并数据
y <- c(A,B)
#数据分组标签
group=as.factor(c(rep(1,10),rep(2,10)))
#载入car包
library(car)
#方差齐性检验
leveneTest(y=y,group = group)
#结果显示,P=0.5505>0.05。说明方差齐性。
Levene's Test for Homogeneity of Variance (center = median)
Df F value Pr(>F)
group 1 0.3703 0.5505
18
#独立样本t检验
t.test(A,B,paired = FALSE)
#结果显示P=0.5639。说明两者没有区别。
Welch Two Sample t-test
data: A and B
t = -0.589, df = 16.463, p-value = 0.5639
alternative hypothesis: true difference in means is not equal to 0
95 percent confidence interval:
-10.100024 5.700024
sample estimates:
mean of x mean of y
83.8 86.0
~~~
**配对样本t检验**
配对样本t检验同样检验两个样本均数及其所代表的总体之间差异是否显著。
独立样本t检验与配对样本t检验同属于双样本t检验,不同点在于配对样本t检验要求两个样本之间存在某些配对关系。
**常见配对关系**:
同一样本两种不同处理方法的检验结果
同一样本前后时间点的检验结果
**适用条件**
正态性
**示例**
有20名女性分为10对,试吃两种药。经过一段时间后,药效如下。问两种药是否有区别
~~~R
#生成数据
drug1 <- c(4.4,5,5.8,4.6,4.9,4.8,6,5.9,4.3,5.1)
drug2 <- c(6.2,5.2,5.5,5,4.4,5.4,5,6.4,5.8,6.2)
#配对样本t检验
t.test(drug1,drug2,paired = TRUE)
#结果显示,P=0.1575>0.05,不能说两者存在显著差别。
Paired t-test
data: drug1 and drug2
t = -1.5417, df = 9, p-value = 0.1575
alternative hypothesis: true difference in means is not equal to 0
95 percent confidence interval:
-1.0609306 0.2009306
sample estimates:
mean of the differences
-0.43
~~~
#### **方差检验**
------
方差分析(analysis of variance ,ANOVA)就是通过检验各总体的均值是否相等来判断分类型自变量对数值型因变量是否有显著影响。
**示例**
我们使用的是R里内置的“npk”数据集,该数据集由24行和5列数据组成,第一列代表区组(共6个),N、P和K分别代表氮、磷和钾元素的使用情况,yield代表豌豆产量,该数据集主要是用来研究不同肥料对豌豆产量的影响。
~~~R
fit <- aov(yield ~ N, data=npk)
fit <- aov(yield ~ N + block, data=npk)
~~~
#### 卡方检验
------
卡方检验就是统计样本的实际观测值与理论推断值之间的偏离程度,实际观测值与理论推断值之间的偏离程度就决定卡方值的大小,卡方值越大,越不符合;卡方值越小,偏差越小,越趋于符合,若两个值完全相等时,卡方值就为0,表明理论值完全符合。
**适用条件**:
1.所有的理论数T≥5并且总样本量n≥40,用Pearson卡方进行检验.
2.如果理论数T<5但T≥1,并且n≥40,用连续性校正的卡方进行检验.
3.如果有理论数T<1或n<40,则用Fisher’s检验.
**示例**:
判断5种品牌啤酒的爱好者有无显著差异:

~~~R
x<-c(210,312,170,85,223)
chisq.test(x)
~~~

~~~R
x<-matrix(c(46,18,6,8),ncol=2,nrow=2)
chisq.test(x)
chisq.test(x)$expected ###查看理论值
fisher.test(x) ##进行fisher检验
~~~
#### **秩和检验**
------
秩和检验是对原假设的非参数检验,在不需要假设两个样本空间都为正态分布的情况下,测试它们的分布是否完全相同。
~~~R
library(stats)
data("mtcars")
wilcox.test(mpg~am,data = mtcars)
~~~
## 几种常用的R包
目前常用差异分析的R包有edgeR、limma、DESeq2

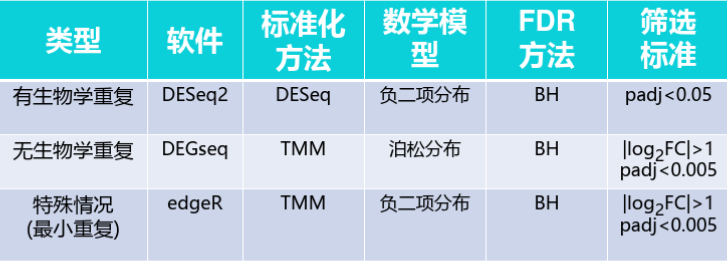
三种包的区别:
1.limma包做差异分析要求数据满足正态分布或近似正态分布,如基因芯片、TPM格式的高通量测序数据。
2.通常认为Count数据不符合正态分布而服从泊松分布。对于count数据来说,用limma包做差异分析,误差较大
3.DESeq2、和 EdgeR都是基于count,然后两个都是NB(negative binomial)但是在估计dispersion parameter的方法上面不一样。
4.limma,edgeR,DESeq2三大包基本是做转录组差异分析的金标准,大多数转录组的文章都是用这三个R包进行差异分析。
5.edgeR差异分析速度快,得到的基因数目比较多,假阳性高(实际不差异,结果差异)。DESeq2差异分析速度慢,得到的基因数目比较少,假阴性高(实际差异,结果不差异)。
6.需要注意的是制作分组信息的因子向量是,因子水平的前后顺序,在R的很多模型中,默认将因子向量的第一个水平看作对照组
如果数据量大并且要求比较conservative的话可以所有方法都用下,然后取并集。
#### 数据预处理
------
~~~R
library("edgeR")
expr = read.csv("mRNA_exprSet.csv",sep = ',',header=T)
head(expr)
expr = avereps(expr[,-1],ID = expr$X) # 对重复基因名取平均表达量,然后将基因名作为行名
expr = expr[rowMeans(expr)>1,] # 去除低表达的基因
library(stringr)
tumor <- colnames(expr)[as.integer(substr(colnames(expr),14,15)) < 10]
normal <- colnames(expr)[as.integer(substr(colnames(expr),14,15)) >= 10]
tumor_sample <- expr[,tumor]
normal_sample <- expr[,normal]
exprSet_by_group <- cbind(tumor_sample,normal_sample)
group_list <- c(rep('tumor',ncol(tumor_sample)),rep('normal',ncol(normal_sample)))
~~~
#### **edgeR**
------
对于`edgeR`的分析流程而言,我们需要输入的数据包括:
1. 表达矩阵(`counts`)
2. 分组信息(`group`)
3. 拟合信息(`design`):指明如何根据样本的分组进行建模
`edgeR`默认使用 trimmed mean of M-values (TMM) 计算文库的scale factor进行normalization,以最大程度地缩小样本间基因表达量的log-fold change。这是因为TMM 法认为样本间大部分的基因都没有发生差异表达,而那些真正差异表达的基因并不会受到normalization的严重影响。如此一来,便将那些由于测序引起的差异表达基因的表达量给校正了,消除了一部分的假阳性。
~~~r
data = exprSet_by_group
group_list = factor(group_list)
design <- model.matrix(~0+group_list)
rownames(design) = colnames(data)
colnames(design) <- levels(group_list)
DGElist <- DGEList( counts = data, group = group_list)
## Counts per Million or Reads per Kilobase per Million
keep_gene <- rowSums( cpm(DGElist) > 1 ) >= 2 ## 自定义
table(keep_gene)
DGElist <- DGElist[ keep_gene, , keep.lib.sizes = FALSE ]
DGElist <- calcNormFactors( DGElist )
DGElist <- estimateGLMCommonDisp(DGElist, design)##为所有基因都计算同样的dispersion
DGElist <- estimateGLMTrendedDisp(DGElist, design)##根据不同基因的均值--方差之间的关系来计算dispersion,并拟合一个trended model
DGElist <- estimateGLMTagwiseDisp(DGElist, design)##计算每个基因的dispersion,并利用经验性贝叶斯方法(empirical bayes)压缩至Trend Didspersion。个人认为这一项相当于GLM中每个基因的beta值
#####################负二项式广义对数线性模型
fit <- glmFit(DGElist, design)
results <- glmLRT(fit, contrast = c(-1, 1))
####################类似然负二项式广义对数线性模型
fit <- glmQLFit(dge, design, robust = TRUE) #拟合模型
lrt <- glmQLFTest(fit)
nrDEG_edgeR <- topTags(results, n = nrow(DGElist))
nrDEG_edgeR <- as.data.frame(nrDEG_edgeR)
head(nrDEG_edgeR)
padj = 0.01 # 自定义
foldChange= 2 # 自定义
nrDEG_edgeR_signif = nrDEG_edgeR[(nrDEG_edgeR$FDR < padj &
(nrDEG_edgeR$logFC>foldChange | nrDEG_edgeR$logFC<(-foldChange))),]
nrDEG_edgeR_signif = nrDEG_edgeR_signif[order(nrDEG_edgeR_signif$logFC),]
~~~
如果没有生物学重复

~~~R
library(edgeR)
##跟DESeq2一样,导入数据,预处理(用了cpm函数)
exprSet<- read.table(file = "test.txt", sep = "\t", header = TRUE, row.names = 1, stringsAsFactors = FALSE)
group_list <- factor(c(rep("Contral",1),rep("Treat",1)))
##设置分组信息,并做TMM标准化
exprSet <- DGEList(counts = exprSet, group = group_list)
bcv = 0.4 #设置BCV值
et <- exactTest(exprSet, dispersion=bcv^2)
write.csv(topTags(et, n = nrow(exprSet$counts)), 'result.csv', quote = FALSE) #输出主要结果
~~~
limma,DEseq2的代码实现可以在https://blog.csdn.net/weixin_43700050/article/details/98085127找到。

参考:
1.https://www.jianshu.com/p/bb0bd72bc428.
2.https://zhuanlan.zhihu.com/p/57756620.
3.https://blog.csdn.net/weixin_43700050/article/details/98085127.
4.https://blog.csdn.net/u010608296/article/details/114135253?utm_medium=distribute.pc_relevant.none-task-blog-2%7Edefault%7EBlogCommendFromMachineLearnPai2%7Edefault-2.control&dist_request_id=1328761.11124.16172439205851981&depth_1-utm_source=distribute.pc_relevant.none-task-blog-2%7Edefault%7EBlogCommendFromMachineLearnPai2%7Edefault-2.control.edgeR/limma/DESeq2
5.https://www.jianshu.com/p/17ae5bee7bb0
6.https://www.jianshu.com/p/517167c50a5f